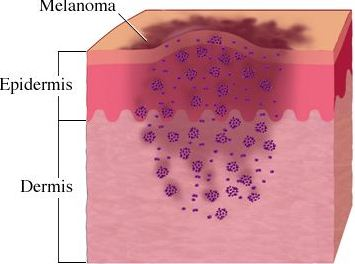
It is well accepted that T cells can recognize and kill tumors that arise in individuals but that tumor cells escape immune surveillance due to the immunosuppressive tumor microenvironment that renders these T cells dysfunctional is less understood. Only a relatively small number of antigens that T cells recognize for tumor-killing have been identified, and the methods used to identify these antigens are quite cumbersome. In a recent article in Nature Medicine, Robbins et al. utilize informatics methods to identify mutated tumor antigens in melanoma patients that allowed effective targeting by anti-tumor T cells.

In an effort to identify clinically relevant mutated tumor cell epitopes recognized by T cells, Robbins et al. first performed whole-exome sequencing of tumor cells and matched normal cells from melanoma patients who demonstrated tumor regression following adoptive transfer of autologous tumor infiltrating lymphocytes (TILs). Mutations in tumor cells that resulted in amino acid changes were identified and then screened using an MHC binding algorithm that predicts high affinity binding of peptide sequences to specific HLA alleles. Candidate peptides of 9-10 amino acids in length were synthesized and pulsed with specific HLA-expressing target cell lines to load the peptides into the MHC complex. Peptide-pulsed target cells or autologous tumor cell lines were then cultured with autologous TILs from the same donor and IFN-gamma production was assessed as a read out of T cell activation.
Three metastatic melanoma patients were assessed using this methodology. The first patient was homozygous for HLA-A*0201, and thus mutated melanoma cell line peptides predicted to bind to the HLA-A*0201 allele were identified by the MHC-binding algorithm. From this donor, 4 out of 55 candidate peptides elicited IFN-gamma responses from autologous T cells cultured with peptide-pulsed target cells. Two of these mutated peptides were found to correspond to the casein kinase1α1 protein (CSNK1A1), one peptide was mapped to the growth arrest specific 7 gene (GAS7) gene, and the fourth was a fragment of the HAUS augmin-like complex, subunit 3 (HAUS3) protein. The wild-type versions of each of these peptides bound very poorly (100-10,000 fold less) or not at all to the HLA and were not recognized by T cells. Two other donors were assessed for predicted binding of mutated peptides to HLA-A*0101 and HLA-A*1101. Autologous T cell responses were found to be activated in response to mutated peptides from pleckstrin homology domain containing, family M member 2 (PLEKHM2), protein phosphatase 1 regulatory subunit 3B (PPP1R3B), matrilin 2 (MATN2), and cyclin-dependent kinase 12 (CDK12) genes, but not their wild-type counterparts. Furthermore, tumor lines were validated to express these mutated proteins.
Finally, the authors compared the reactivity of peripheral blood mononuclear cells (PBMCs) drawn before and after adoptive TIL transfer into two of these patients to determine if anti-tumor reactive T cell clones persisted in vivo. T cells that recognized the same tumor antigens as the TILs were identified post-adoptive transfer at greater levels than prior to adoptive transfer. Thus, T cells that recognize mutated tumor epitopes may play a clinically relevant role in mediating tumor regression. Many questions remain, including a direct demonstration that such tumor-reactive TILs are responsible for mediating the observed tumor regression in these patients, and whether further mutation of these residues might facilitate immune escape later it the course of disease.
Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Robbins PF, Lu YC, El-Gamil M, Li YF, Gross C, Gartner J, Lin JC, Teer JK, Cliften P, Tycksen E, Samuels Y, Rosenberg SA. Nat Med. 2013 May 5. doi: 10.1038/nm.3161.
NetMHCpan, a method for quantitative predictions of peptide binding to any HLA-A and -B locus protein of known sequence. Nielsen M, Lundegaard C, Blicher T, Lamberth K, Harndahl M, Justesen S, Røder G, Peters B, Sette A, Lund O, Buus S. PLoS One. 2007 Aug 29;2(8):e796.


 With the increasing prevalence of neurodegenerative disorders in the aging population, it has become more and more important to understand the molecular pathways that regulate and advance these disorders. Due to the high level of complexity of the mammalian brain, it is very difficult to devise improved targeted treatments. The biggest limitation in neurodegenerative disease research being the lack of viable biomarkers for the elder population. Neurodegenerative disorders such as Alzheimer’s, Parkinson’s and polyglutamine diseases, share many pathogenic abnormalities such as the accumulation of misfolded proteins due to mutations rendering them resistant to degradation or over-expression of the wild type form.
With the increasing prevalence of neurodegenerative disorders in the aging population, it has become more and more important to understand the molecular pathways that regulate and advance these disorders. Due to the high level of complexity of the mammalian brain, it is very difficult to devise improved targeted treatments. The biggest limitation in neurodegenerative disease research being the lack of viable biomarkers for the elder population. Neurodegenerative disorders such as Alzheimer’s, Parkinson’s and polyglutamine diseases, share many pathogenic abnormalities such as the accumulation of misfolded proteins due to mutations rendering them resistant to degradation or over-expression of the wild type form.