
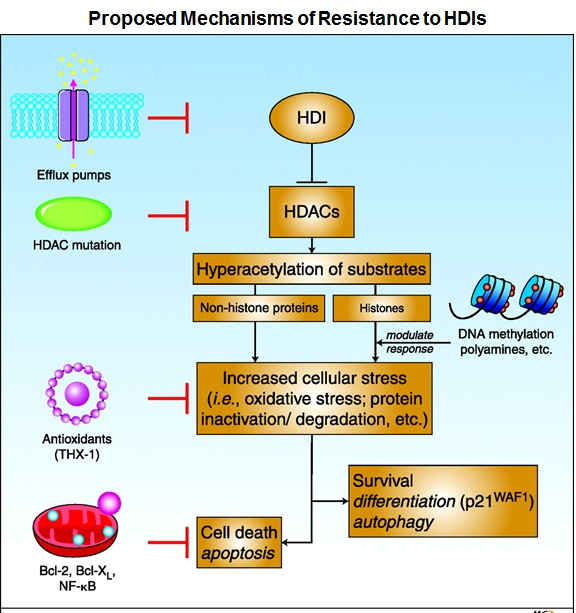
Histone deacetylase (HDAC) inhibitors (HDIs), a new class of epigenetic anti-tumor agents, have shown promise so far in the treatment of hematologic malignancies. Many, if not most, cell lines tested in vitro show sensitivity to HDIs, and synergy in combinations is also often noted. Yet, Phase I and II trials of HDIs have uniformly been disappointing in solid tumors. Even preclinical models in which HDIs exhibited potent anti-tumor effects in vivo have not succeeded in the clinic. Therefore, a detailed understanding of the mechanisms of resistance to HDIs may lead to strategies designed to increase clinical efficacy. Studies have proposed several mechanisms of resistance which include increased expression of the P-glycoprotein (Pgp) encoding multidrug-resistance gene ABCB1, increased express ion of reactive oxygen species (ROS) scavenger protein thioredoxin, elevated expression of anti-apoptotic proteins Bcl-2 and Bcl-xL, increased expression of histone HDAC enzymes, and activation of several signaling pathways including MAPK, phosphoinositide 3-kinase and signal transducer and activator of transcription.
ion of reactive oxygen species (ROS) scavenger protein thioredoxin, elevated expression of anti-apoptotic proteins Bcl-2 and Bcl-xL, increased expression of histone HDAC enzymes, and activation of several signaling pathways including MAPK, phosphoinositide 3-kinase and signal transducer and activator of transcription.
The FDA approved histone deacetylase inhibitor romidepsin (istodax®) for the treatment of cutaneous T-cell lymphoma (CTCL) in 2009 and for the treatment of peripheral T-cell lymphoma (PTCL) in 2011. Disease progression was noted in some patients who initially responded to therapy, while disease in other patients did not respond to therapy suggesting that both de novo and acquired resistance to romidepsin were observed during the trial. Thus, studies are needed to determine the mechanisms of resistance to the HDI romidepsin particularly in T-cell lymphoma.
Several studies have noted selection of Pgp overexpression in vitro as a mechanism of drug resistance to romidepsin. The ease of selection of Pgp is in part because romidepsin is a substrate for Pgp, and in part based on ABCB1 gene induction as a consistent cellular response to HDIs. While induction of ABCB1 has been noted in normal and malignant peripheral blood mononuclear cells of patients treated with romidepsin, no evidence for Pgp-mediated resistance emerged in clinical samples obtained at the time of disease progression from patients with CTCL or PTCL. Therefore, identification of non-Pgp resistance mechanisms for romidepsin is warranted. A recent study published in the peer reviewed journal Blood by Chakraborty et al. (Blood. 2013 Mar 26) reported that activation of the mitogen activated protein kinase (MAPK) pathway conferred resistance to romidepsin through degradation of the pro-apoptotic BH-3 only protein Bim. To explore Pgp-independent mechanisms of resistance to romidpesin, a CTCL model consisting of the HuT78 cell line and its romidepsin-selected sublines were used. These sublines were separately selected in romidepsin in the presence of the Pgp-inhibitors verapamil or valspodar (PSC833) to avoid overexpression of Pgp. The resulting cell lines were resistant to romidepsin and inhibition of Pgp could reverse the resistance of only the cells that had not been selected in the presence of the Pgp inhibitors. The failure to reverse the resistance of the romidepsin-selected cells with a Pgp inhibitor suggested a different mechanism of action, which had not been identified in the earlier studies. A gene microarray study detected increased expression of the insulin receptor in the romidepsin-selected cells compared to the parental HuT78 cells. In addition, romidepsin-resistant cells also exhibited increased activation of MEK protein, a downstream component of the MAPK signaling pathway. Treating these resistant cells with the allosteric MEK inhibitors resulted in exquisite sensitivity while the parental HuT78 cells did not respond to the MEK inhibition. Restoration of the pro-apoptotic protein Bim was noted in romidepsin-resistant cells following MEK inhibition which was otherwise found to be degraded in the resistant cells. Combined treatment of MEK inhibitor with romidepsin also caused increased death of resistant cells. In their study, Chakraborty and colleagues also noted loss of Bim in the skin biopsy samples obtained from CTCL patients who experienced disease progression after romidepsin treatment. In addition, this study also reported perturbation of the MAPK regulated genes in the CTCL patients treated with romidepsin. Collectively these observations suggested that activation of the MAPK pathway may limit efficacy of the HDI romidepsin through degradation of the pro-apoptotic protein Bim, and in future clinical trials combination of romidepsin with MEK inhibitor may exhibit promising results.
References:
1.Fantin VR, Richon VM. Mechanisms of resistance to histone deacetylase inhibitors and their therapeutic implications. Clin Cancer Res. 2007;13(24):7237-7242.
2.Chakraborty AR, Robey RW, Luchenko VL, et al. MAPK pathway activation leads to Bim loss and histone deacetylase inhibitor resistance: rationale to combine romidepsin with a MEK inhibitor. Blood. 2013.