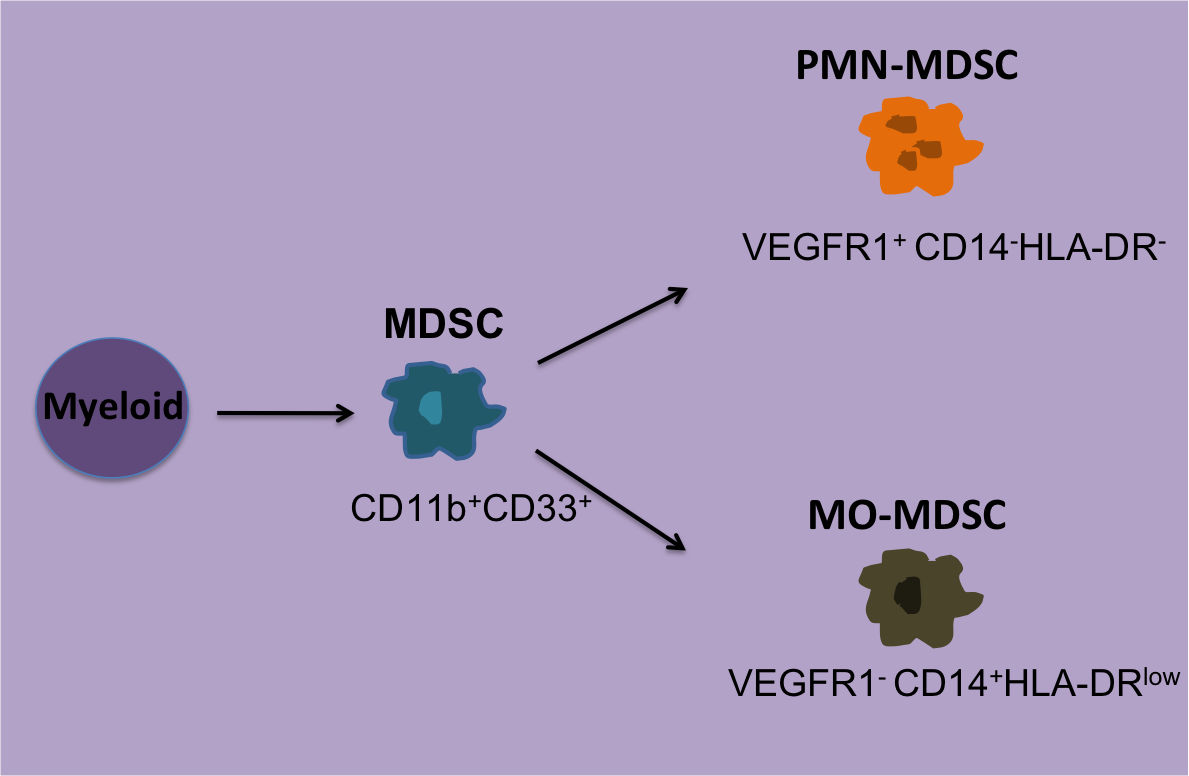
Immunomonitoring of T cell based immune responses spans a wide range of therapeutic applications such as infectious and autoimmune diseases and is particularly important for vaccine research. Regardless of the therapeutic application, immunomonitoring can be a daunting task due to the variability of methods and protocols available. There are several commonly used functional assays for the enumeration of antigen specific CD8+ T cells and there is great variability in the protocols that are used for these assays. Thus, making it increasing difficult to thoroughly interpret data obtained from multi-center clinical trials and to compare results between laboratories. In order to address some of the issues associated with immunomonitoring of clinical trials, the Association for Immunotherapy of Cancer (CIMT) formed a CIMT monitoring panel tasked to standardize protocols for assaying T cell antigen immune responses. Thirteen centers from 6 different European countries participated in this study. They were given the same samples and asked to determine the number of antigen specific T cells and assess their antigen specific function using tetramer staining and a functional assay of their choice. Common techniques used for monitoring antigen induced immune responses included ELISPOT assays, HLA-multimer staining and intracellular cytokine staining (ICS).
Pre-tested samples of peripheral blood mononuclear cells (PBMC), synthetic peptides, and PE-conjugated HLA-tetramers were distributed to each center. Using HLA-typed healthy volunteers, PBMCs were isolated by Ficoll density gradient separation. Each sample was tested for T cell reactivity against CMV and influenza. All centers received an HLA-A negative control as well as HLA-A positive samples consisting of a combination of CMV and influenza reactive PBMCs. The study comprised of 2 phases; Phase I consisted of all centers performing the assays with their commonly used protocols, and in Phase II each center received optimized protocols based on the findings from Phase I.
For Phase I’s tetramer-staining assay, the laboratories could choose to stain samples with antibodies (Ab) for CD8+ alone, CD3+CD8+, or CD4+ CD8+ and use their preferred Ab clone, fluorescent dye, and Ab concentration. For the functional assays synthetic peptides were provided and each group could choose either the INF-γ ELISPOT assay, FACS-based intracellular INF-γ staining or both with their antigen concentration of choice ranging from 1-10 g/ml. To reduce variability in FACS analysis, sample plots were provided as well as gate settings and quadrants. Tetramer-staining data reported included; number of viable cells post-thawing, cytometer model, number of lymphocytes and/or CD8+ cells analyzed. Data was presented as percent of tetramer-positive cells among CD8+, CD3+CD8+, or CD4+ lymphocytes depending on what antibody cocktail was chosen. For the functional assays each center reported the type of ELISPOT plates used, reagents and conditions used, and number cells tested.
Tetramer results from the Phase I study showed the number CD8+ cells analyzed significantly affected the sensitivity of tetramer staining. Antigen-specific T cell reactivity when less than 30,000 CD8+ T cells were counted resulted in only 70% responsiveness detected. In contrast, when more than 30,000 CD8+ cells were counted, an 89% response was observed. Although, when antigen-specific T cells were present at high frequencies the number of counted cells did not matter. Interestingly, Ab clone variability, Ab concentration, or cytometer type did not result in any significant differences. Thus, the main factors affecting antigen-specific T cell reactivity by tetramer staining is the number of CD8+ cells used. For Phase II it was then recommended at least 1 x106 PBMCs are used for this assay.
The majority of groups chose the INF-γ ELISPOTas their functional assay. Results showed a large amount of heterogeneity between the centers. Some centers included a resting phase after thawing the cells, of 2-20 hours, resulting in 73% positive reactivity (number of spot forming cells per seeded PBMC). In contrast, not allowing a resting phase resulted in only detecting 30% of the positive cells. Additionally, intra-center replicate reproducibility was significantly affected by the number of replicates used, where duplicates often failed the Student t test and triplicates were sufficient to reach statistical significance. Addition of allogenic-APCs for binging and presentation of the synthetic peptides was found to have a negative effect on detection response (28% of all responses vs. 58%). When looking at the number of cells seeded per well, those with more than 4 x105 PBMC detected 71% positive samples and those with less than 4 x105 only detected 43%. Granted, when antigen specific T cells were available at high frequencies the number of counted cells did not affect the response rates. Consequently, Phase II’s minimum requirements for the INF-γ ELISPOT protocol included: (1) triplicates should be performed for each test antigen (2) avoid using allogenic-APCs (3) include a resting phase (4) use over 4 x 105 PBMCs per well.
Another interesting finding from this study was that lab experience in performing these assays had no effect on the performance of the assays compared to labs that had just adopted the techniques. Further highlighting the importance of developing standardized protocols for immunomonitoring assays. This study did not however, address specific detection limits for the ELISPOT assays, the variability between ELISPOT plate readers, nor serum source effects on background and specificity. In addition, it was not reported whether live/dead cell stains where included in the tetramer assays and how combinations of these may have had an effect on the sensitivity of the assay.
Overall, this study identified several factors that should be generally implemented when performing tetramer staining and INF-γ ELISPOT assays with cryopreserved PBMC samples. Furthermore, these protocol modifications are particularly important when assaying antigen-specific T cell populations present at low frequencies.
Reference:
The CIMT-monitoring panel: a two-step approach to harmonize the enumeration of antigen-specific CD8+ T lymphocytes by structural and functional assays. Britten CM, Gouttefangeas C, Welters MJ, Pawelec G, Koch S, Ottensmeier C, Mander A, Walter S, Paschen A, Müller-Berghaus J, Haas I, Mackensen A, Køllgaard T, thor Straten P, Schmitt M, Giannopoulos K, Maier R, Veelken H, Bertinetti C, Konur A, Huber C, Stevanović S, Wölfel T, van der Burg SH. Cancer Immunol Immunother. 2008 Mar;57(3):289-302. Epub 2007 Aug 25.



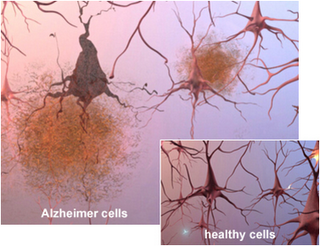
 With the increasing prevalence of neurodegenerative disorders in the aging population, it has become more and more important to understand the molecular pathways that regulate and advance these disorders. Due to the high level of complexity of the mammalian brain, it is very difficult to devise improved targeted treatments. The biggest limitation in neurodegenerative disease research being the lack of viable biomarkers for the elder population. Neurodegenerative disorders such as Alzheimer’s, Parkinson’s and polyglutamine diseases, share many pathogenic abnormalities such as the accumulation of misfolded proteins due to mutations rendering them resistant to degradation or over-expression of the wild type form.
With the increasing prevalence of neurodegenerative disorders in the aging population, it has become more and more important to understand the molecular pathways that regulate and advance these disorders. Due to the high level of complexity of the mammalian brain, it is very difficult to devise improved targeted treatments. The biggest limitation in neurodegenerative disease research being the lack of viable biomarkers for the elder population. Neurodegenerative disorders such as Alzheimer’s, Parkinson’s and polyglutamine diseases, share many pathogenic abnormalities such as the accumulation of misfolded proteins due to mutations rendering them resistant to degradation or over-expression of the wild type form.