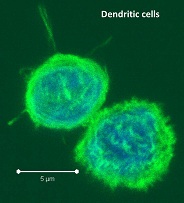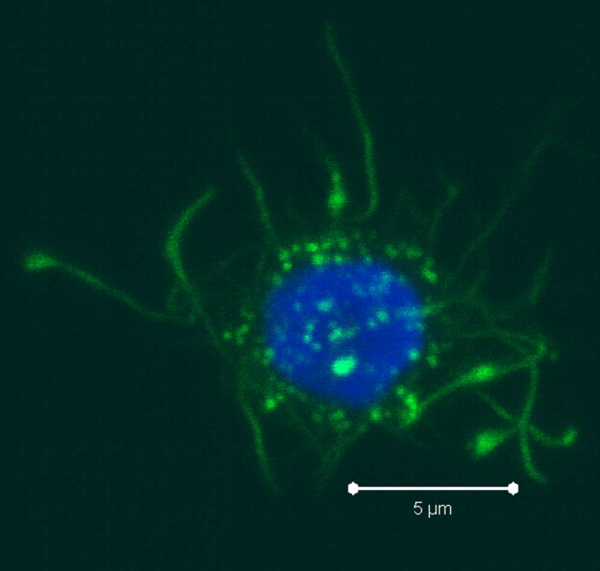
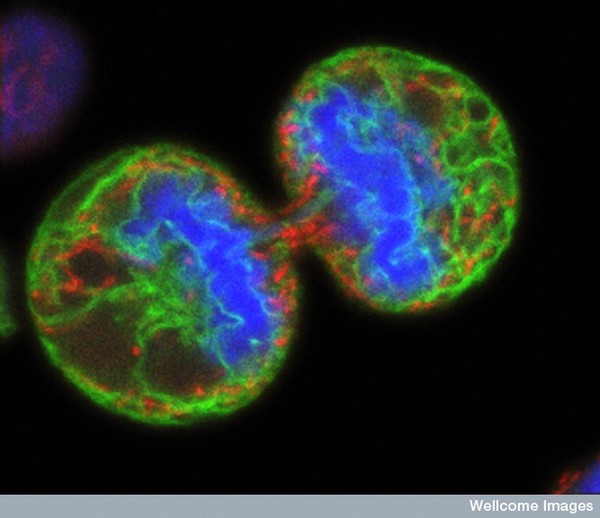
 PBMCs are not just a source of many different circulating immune cell types, but also a source of potential cells that one can generate in vitro. One excellent and long-standing example of this is the generation of dendritic cells (DCs) from monocytes. Monocyte derived DCs (mDCs) are an excellent tool for researchers to do immunological assays requiring a source of professional antigen presenting cells (APCs). While circulating B cells are capable of antigen presentation and T cell activation, they do not offer the robust response that DCs do. The generation of mDCs is a relatively simple protocol that anyone can do with just a source of PBMCs, a few important cytokines, and, of course, some media and incubator space. After this protocol, you will have obtained immature mDCs that can then be matured for use as APCs in your assay.
PBMCs are not just a source of many different circulating immune cell types, but also a source of potential cells that one can generate in vitro. One excellent and long-standing example of this is the generation of dendritic cells (DCs) from monocytes. Monocyte derived DCs (mDCs) are an excellent tool for researchers to do immunological assays requiring a source of professional antigen presenting cells (APCs). While circulating B cells are capable of antigen presentation and T cell activation, they do not offer the robust response that DCs do. The generation of mDCs is a relatively simple protocol that anyone can do with just a source of PBMCs, a few important cytokines, and, of course, some media and incubator space. After this protocol, you will have obtained immature mDCs that can then be matured for use as APCs in your assay.
The first step in generating mDCs is to decide how you would like to isolate the monocyte population from your PBMCs, which serve as your precursor cells for DCs. The easiest and cheapest way is to simply plate your PBMCs on a cell culture dish and let the inherent qualities of monocytes go to work. Monocytes are unique amongst other PBMC cells in their tendency to stick to plastic. An incubation period between 1-24 hours will allow your monocytes to adhere to the dish and let you gently wash off any other PBMCs. The alternative to the adherence method for isolating monocytes is to use a magnetic antibody based technology of your choice. Several companies, such as Miltenyi Biotec, Life Technologies, and Stem Cell Technologies, offer excellent kits for this. While the adherence method is cheaper, antibody based kits give you higher monocyte recovery and purity, which may or may not matter depending on your downstream assays.
Once you have your monocytes isolated from your PBMCs, you can begin the 7 day culture to generate mDCs. Monocytes can be plated in a standard cell culture media along with two important cytokines, GM-CSF and IL-4 (50ng/mL and 100ng/mL). GM-CSF will push the monocytes down a DC differentiation pathway. IL-4 will inhibit the monocytes from differentiating into macrophages, thereby insuring they become DCs. Continue the culture for 6-8 days and be sure to refresh your cytokines every other day.
As the monocytes differentiate over the culture period, note their progress by examining them with your tissue culture room microscope. The cells should appear as fairly round and are generally 2-3 times the size of lymphocytes. It is important to note that the mDCs will not appear like the elongated cartoon DCs with long extensions you see in text books. Those DC characteristics are generally only found in tissues and not in vitro. While you may see some cells that resemble this, those are more likely to be somewhat of a natural stromal layer, made up of cells including macrophages, that the monocyte culture develops to support cell growth. In fact, the immature mDCs will have very few if any, cytoplasmic protrusions.
 Once the culture period has finished, between 6-8 days, the mDCs can be collected. The exact day is not critical, as long as you remain consistent in the day you pick for your following experiments. To collect the mDCs, gently wash the culture dishes with several streams of media by pipetting up and down. The mDCs, which are currently immature, will be somewhat floating and only loosely adherent. Because of their loose adherence, they require several rounds of gentle pipetting, but do not require cell scraping, EDTA, or trypsin treatment. Note that the culture dishes will still contain some adherent cells. Do not worry about these cells, since these are not the loosely adherent DCs we are interested in.
Once the culture period has finished, between 6-8 days, the mDCs can be collected. The exact day is not critical, as long as you remain consistent in the day you pick for your following experiments. To collect the mDCs, gently wash the culture dishes with several streams of media by pipetting up and down. The mDCs, which are currently immature, will be somewhat floating and only loosely adherent. Because of their loose adherence, they require several rounds of gentle pipetting, but do not require cell scraping, EDTA, or trypsin treatment. Note that the culture dishes will still contain some adherent cells. Do not worry about these cells, since these are not the loosely adherent DCs we are interested in.
After completion of these steps, you should have a nice population of immature mDCs, which express CD11c, CD1c, and are CD123-. In my next post, I will cover some tips and tricks for analyzing these cells by flow cytometry. Importantly, I will also cover ways to mature the immature mDCs for use as APCs.
 Colt Egelston is currently a post-doctoral fellow at the Beckman Research Institute of the City of Hope, in Duarte, CA. He received his Ph.D. from Rush University in Chicago and is interested in all things immunology.
Colt Egelston is currently a post-doctoral fellow at the Beckman Research Institute of the City of Hope, in Duarte, CA. He received his Ph.D. from Rush University in Chicago and is interested in all things immunology.


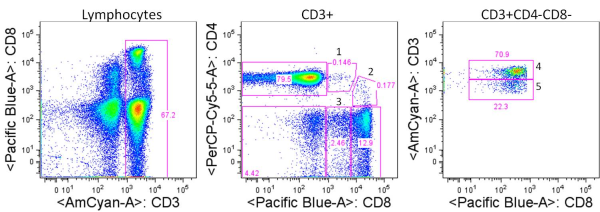
 There are many strategies for assessing the function of T cells from
There are many strategies for assessing the function of T cells from 


 96-well Plates: Different types of 96-well plates are available for different assay types. There are various surface coatings including tissue-culture treated polystyrene for cell cultures, uncoated, and others. Plates can have various plate bottom geometries and optical characteristics. For instance there are black plates available for light-sensitive assays. For protocols involving volumes larger then 250ul, there are deep-well plates that carry a 2ml volume per well.
96-well Plates: Different types of 96-well plates are available for different assay types. There are various surface coatings including tissue-culture treated polystyrene for cell cultures, uncoated, and others. Plates can have various plate bottom geometries and optical characteristics. For instance there are black plates available for light-sensitive assays. For protocols involving volumes larger then 250ul, there are deep-well plates that carry a 2ml volume per well. Multichannel Vacuums: Companies such as V&P Scientific offer a multitude of multichannel vacuum manifolds that fit plates of different depths for removing supernatant from wells via vacuum apparatus. Often these will be the proper length such that they don’t touch the well bottom and work well with removing buffers from centrifuged PBMC cell cultures, such as during washing steps for flow-cytometry.
Multichannel Vacuums: Companies such as V&P Scientific offer a multitude of multichannel vacuum manifolds that fit plates of different depths for removing supernatant from wells via vacuum apparatus. Often these will be the proper length such that they don’t touch the well bottom and work well with removing buffers from centrifuged PBMC cell cultures, such as during washing steps for flow-cytometry.