
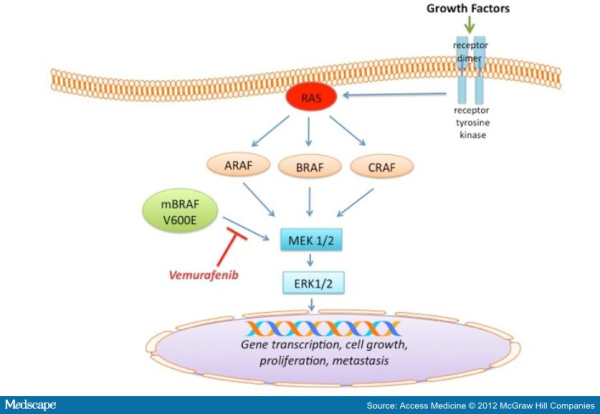
With the increasing knowledge about the role of V600E B-RAF mutation in melanoma progression, efforts have been made to target and inhibit this kinase and its downstream signaling. The ATP-competitive type I B-RAF inhibitors vemurafenib and dabrafenib (GSK2118436) exhibit remarkable anti-cancer activity in patients with V600E B-RAF mutant melanomas. Targeted inhibition of BRAF with vemurafenib causes tumor regression and extends survival in many patients with BRAF-mutant metastatic melanoma. In 2011, the Food and Drug Administration (FDA) approved vemurafenib tablets (ZELBORAF) for the treatment of patients with unresectable or metastatic melanoma with the V600EBRAF mutation. Vemurafenib is not recommended for use in patients with wild-type BRAF melanoma. Even though a very high percentage of patients respond to vemurafenib, resistance to this drug develops relatively quickly. With continued treatment, the emergence of resistance can be seen as soon as 6-8 weeks following initial documentation of response. However, a subset of patients maintains drug responsiveness beyond 18 months. Overall, the median duration of responsiveness to vemurafenib is 8 months. Some studies described involvement of certain molecular mechanisms associated with vemurafenib resistance in melanoma. Reactivation of the MAPK pathway, NRAS mutation, overexpression of platelet derived growth factor beta receptor, activation of PI3K/AKT signaling, genomic amplification of V600EBRAF are some of the m echanisms of acquired resistance to BRAF inhibitors (for detail please refer to my blog post titled ” RESISTANCE TO B-RAF INHIBITORS IN MELANOMA’’). Due to heterogeneous nature of cancer cells, it is crucial to gain a thorough understanding of the underlying drug-resistance mechanisms so that we can develop novel strategies to circumvent resistance and achieve more-prolonged responses. A recent study published in the journal Nature by Das Thakur and colleagues reported that intermittent treatment of vemurafenib prevented resistance in primary human melanoma xenografts. To study mechanisms of resistance to vemurafenib, Das Thakur et al. developed an animal model by continuously treating mice bearing a vemurafenib-naive, patient-derived BRAF-mutant melanoma with vemurafenib until drug resistance developed. Exome sequence analysis did not detect any secondary mutations in the coding sequences of BRAF, NRAS, KRAS, HRAS, and MEK1 in the resistant tumors. No alternatively spliced isoform of V600EBRAF, another known mechanism of vemurafenib resistance in melanoma was also detected in the resistant tumors. However, increased expression of V600EBRAF protein was noted in the resistant tumors and inhibition of V600EBRAF gene by RNA interference resulted in suppression of proliferation. These data suggested that the tumor cells were BRAF oncogene dependent and the observed drug resistance was due to the increased expression of V600EBRAF protein. In addition to these, another interesting observation was noted in this study when Das Thakur et al. tried to establish cell lines derived from the drug-resistant tumors. Cell lines derived from the drug-resistant tumors could not be developed without vemurafenib, where withdrawal of vemurafenib from the newly established cell lines changed cell morphology and decreased proliferation. This suggested that vemurafenib-resistant tumor cells in melanoma suffer a fitness deficit in the absence of vemurafenib. A similar type of vemurafenib dependency was also observed in SK-MEL239-C3 melanoma cells in which resistance is due to expression of a splice variant of V600EBRAF, and also in tumor cells isolated from a BRAF-mutated vemurafenib-resistant melanoma patient. Consistent with these findings, Das Thakur et al. observed tumor regression within 10 days in mice bearing vemurafenib resistant melanoma following cessation of vemurafenib treatment, although tumors eventually started re-growing. Collectively these results suggested that withdrawal of vemurafenib might create a hostile environment for drug-resistant cells and detain the onset of drug resistance. A comparison study made between continuous and intermittent vemurafenib treatment in human melanoma xenografts bearing mice further validated these observations. Drug resistance was developed in mice with 100 days receiving continuous treatment, whereas none of the mice on the intermittent treatment schedule exhibited drug resistance after 200 days of treatment. Therefore, these findings recommend that discontinuous treatment of vemurafenib may select against drug-resistant cells and prolong the responses to vemurafenib in melanoma. Future studies are needed especially in clinical trials to validate this proposal.
echanisms of acquired resistance to BRAF inhibitors (for detail please refer to my blog post titled ” RESISTANCE TO B-RAF INHIBITORS IN MELANOMA’’). Due to heterogeneous nature of cancer cells, it is crucial to gain a thorough understanding of the underlying drug-resistance mechanisms so that we can develop novel strategies to circumvent resistance and achieve more-prolonged responses. A recent study published in the journal Nature by Das Thakur and colleagues reported that intermittent treatment of vemurafenib prevented resistance in primary human melanoma xenografts. To study mechanisms of resistance to vemurafenib, Das Thakur et al. developed an animal model by continuously treating mice bearing a vemurafenib-naive, patient-derived BRAF-mutant melanoma with vemurafenib until drug resistance developed. Exome sequence analysis did not detect any secondary mutations in the coding sequences of BRAF, NRAS, KRAS, HRAS, and MEK1 in the resistant tumors. No alternatively spliced isoform of V600EBRAF, another known mechanism of vemurafenib resistance in melanoma was also detected in the resistant tumors. However, increased expression of V600EBRAF protein was noted in the resistant tumors and inhibition of V600EBRAF gene by RNA interference resulted in suppression of proliferation. These data suggested that the tumor cells were BRAF oncogene dependent and the observed drug resistance was due to the increased expression of V600EBRAF protein. In addition to these, another interesting observation was noted in this study when Das Thakur et al. tried to establish cell lines derived from the drug-resistant tumors. Cell lines derived from the drug-resistant tumors could not be developed without vemurafenib, where withdrawal of vemurafenib from the newly established cell lines changed cell morphology and decreased proliferation. This suggested that vemurafenib-resistant tumor cells in melanoma suffer a fitness deficit in the absence of vemurafenib. A similar type of vemurafenib dependency was also observed in SK-MEL239-C3 melanoma cells in which resistance is due to expression of a splice variant of V600EBRAF, and also in tumor cells isolated from a BRAF-mutated vemurafenib-resistant melanoma patient. Consistent with these findings, Das Thakur et al. observed tumor regression within 10 days in mice bearing vemurafenib resistant melanoma following cessation of vemurafenib treatment, although tumors eventually started re-growing. Collectively these results suggested that withdrawal of vemurafenib might create a hostile environment for drug-resistant cells and detain the onset of drug resistance. A comparison study made between continuous and intermittent vemurafenib treatment in human melanoma xenografts bearing mice further validated these observations. Drug resistance was developed in mice with 100 days receiving continuous treatment, whereas none of the mice on the intermittent treatment schedule exhibited drug resistance after 200 days of treatment. Therefore, these findings recommend that discontinuous treatment of vemurafenib may select against drug-resistant cells and prolong the responses to vemurafenib in melanoma. Future studies are needed especially in clinical trials to validate this proposal.
References:
1. Das Thakur M, Salangsang F, Landman AS, et al. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature. 2013;494(7436):251-255.
2. Sullivan RJ, Flaherty KT. Resistance to BRAF-targeted therapy in melanoma. Eur J Cancer. 2013;49(6):1297-1304.