
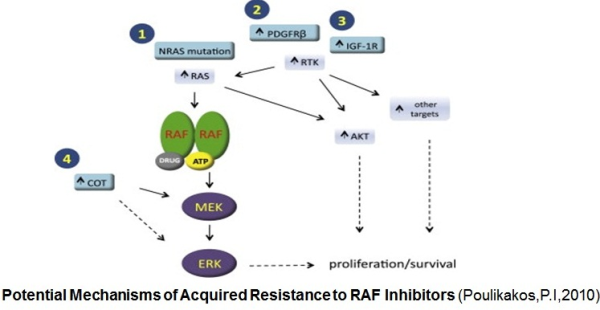
B-RAF is a serine/therionine protein kinase which activates the mitogen-activated protein kinase (MAPK) signaling pathway. As suggested by Hanahan and Weinberg (2000) six major hallmarks of cancer are: independence of proliferation signals, evasion of apoptosis, insensitivity to anti-growth signals, unlimited replicative potential, the ability to invade and metastasize, and induction of angiogenesis for nutrient supply. Abnormalities in the MAPK signaling impinge on most, if not all these processes, and play a critical role in the development and progression of cancer. Activating mutations in B-RAF kinase (mostly V600E B-RAF) are observed in about 50% of melanomas resulting in constitutive activation of the MAPK pathway. With the increasing knowledge about the role of V600E B-RAF mutation in melanoma progression, efforts have been made to target and inhibit this kinase and its downstream signaling. The ATP-competitive type I B-RAF inhibitors vemurafenib and dabrafenib (GSK2118436) exhibit remarkable anti-cancer activity in patients with V600E B-RAF mutant melanomas. However, acquisition of drug resistance virtually occurs in all patients treated with B-RAF inhibitors. In clinical trials with the B-RAF inhibitors, disease progression was noted in most of the patients after 6-7 months of initial response. Investigations at the molecular level suggest that development of resistance is associated with the acquisition of the secondary mutations within the kinase ATP binding site that inhibited the binding of drug to the hydrophobic pocket at so-called “gatekeeper” residue. In a preclinical study Whittaker et al. (2010) reported that a gatekeeper mutation in BRAF at Threonine-259 (T259) residue conferred resistance to the B-RAF inhibitors SB590885 and PLX4720. However, no such mutation was observed in patients whose disease progressed following vemurafenib treatment. This suggests existence of V600EB-RAF-bypass mechanism for acquired resistance to the B-RAF inh ibitors. Activation of the MAPK signaling and increased expression of C-RAF (an isoform of B-RAF) was noted in melanoma cells resistant to B-RAF inhibitors. In this study Villanueva et al. (2010 ) also observed constitutive activation of the insulin-like growth factor receptor 1 (IFGR1), a receptor tyrosine kinase (RTK) in the resistant cells. As IGFR1 activates PI3K/Akt signaling, combined treatment of PI3K and MEK inhibitors resulted in resistance reversal. Increased levels of IGFR1 was also observed in melanoma patients failing vemurafenib suggesting activation of PI3K/Akt signaling via IGFR1 could limit the efficacy of B-RAF inhibitors in the clinic. Up-regulation of other RTKs was also found to be associated with the acquired resistance to vemurafenib. Tumor biopsies of melanoma patients failing vemurafenib exhibited over-expression of the platelet derived growth factor receptor-β (Nazarian et al., 2010).
ibitors. Activation of the MAPK signaling and increased expression of C-RAF (an isoform of B-RAF) was noted in melanoma cells resistant to B-RAF inhibitors. In this study Villanueva et al. (2010 ) also observed constitutive activation of the insulin-like growth factor receptor 1 (IFGR1), a receptor tyrosine kinase (RTK) in the resistant cells. As IGFR1 activates PI3K/Akt signaling, combined treatment of PI3K and MEK inhibitors resulted in resistance reversal. Increased levels of IGFR1 was also observed in melanoma patients failing vemurafenib suggesting activation of PI3K/Akt signaling via IGFR1 could limit the efficacy of B-RAF inhibitors in the clinic. Up-regulation of other RTKs was also found to be associated with the acquired resistance to vemurafenib. Tumor biopsies of melanoma patients failing vemurafenib exhibited over-expression of the platelet derived growth factor receptor-β (Nazarian et al., 2010).
In addition to the increase RTKs activity, resistance to the B-RAF inhibitors was noted to be mediated by genetic alteration in the MAPK signaling pathway. Genetic analysis detected an activating mutation in NRAS in codon 61 in tumor biopsies from patients treated with vemurafenib (Nazarian et al., 2010). Next, there is also evidence of B-RAF alterations in tumor samples collected from patients whose cancer progressed after initial response to B-RAF inhibitors. A study by Shi et al. (2012) reported overexpression of V600EB-RAF as a result of genomic copy-numbers gain in 20% vemurafenib resistant melanoma patients. In the in vitro study, Shi et al. observed restoration of the sensitivity of the B-RAF amplification driven vemurafenib resistant cells to vemurafenib following treatment with the MEK inhibitor selumetinib (AZD6244). This observation provides evidence of the MAPK pathway reactivation as a mechanism of resistance. Existence of a structural changes in B-RAF could also confer resistance as identified by Poulikakos et al. (2011). Their study discovered a splice variant of B-RAF(V600E). Expression of a 61-kDa B-RAF variant lacking RAS-binding domain was identified in the tumors of 32% patients with acquired resistance to vemurafenib.
One of the challenges in melanoma treatment is how to develop effective strategies for overcoming intrinsic and acquired drug resistance to small molecule B-RAF inhibitors. Although the resistance mechanisms identified so far are diverse, most seem to rely upon the reactivation of the MAPK signaling pathway and enhanced signaling output through the PI3K/Akt signaling pathway. Therefore, dual BRAF and PI3K/Akt signaling pathway inhibition may prevent or delay the onset of resistance in melanoma.
References:
1. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. Vol. 100. United States; 2000:57-70.
2. Whittaker S, Kirk R, Hayward R, et al. Gatekeeper mutations mediate resistance to BRAF-targeted therapies. Sci Transl Med. Vol. 2. United States; 2010:35ra41.
3. Villanueva J, Vultur A, Lee JT, et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell. Vol. 18. United States: 2010 Elsevier Inc; 2010:683-695.
4. Nazarian R, Shi H, Wang Q, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. Vol. 468. England; 2010:973-977.
5. Shi H, Moriceau G, Kong X, et al. Melanoma whole-exome sequencing identifies (V600E)B-RAF amplification-mediated acquired B-RAF inhibitor resistance. Nat Commun. Vol. 3. England; 2012:724.
6. Alas S, Bonavida B. Inhibition of constitutive STAT3 activity sensitizes resistant non-Hodgkin’s lymphoma and multiple myeloma to chemotherapeutic drug-mediated apoptosis. Clin Cancer Res. 2003;9(1):316-326.