
The immune privilege of the eye is a widely recognized but frequently oversimplified concept. The notion that the eye possessed unusual immunological characteristics was recognized in the 19th century by van Dooremaal, who observed prolonged survival of murine skin grafts transplanted into the anterior chamber (AC) of the dog eye. The term ocular ‘immune privilege’ was articulated by Medawar, who recognized that the extended survival of foreign grafts in the AC was a remarkable departure from the fate of similar grafts transplanted to sites outside of the eye. Almost 30 years later, the seminal studies of Kaplan et al. demonstrated that alloantigenic cells introduced into the AC in fact did escape from the eye and induced a deviant immune response in which serum alloantibodies were generated, while systemic cell-mediated immune responses were suppressed in an antigen-specific manner. Subsequent studies in mice confirmed this AC-associated immune deviation (ACAID) and demonstrated that it is an important contributor to the immune privilege of the eye.

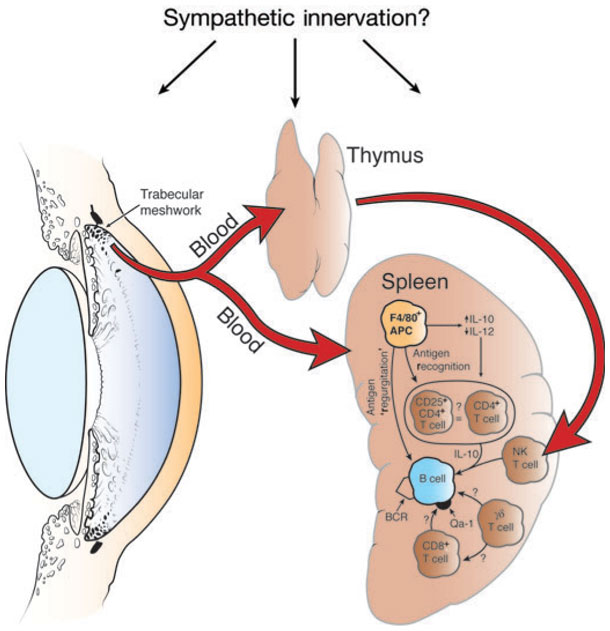
Figure 1. Organ systems involved in the induction of ACAID.
A brief description of the complex cellular-interplay, that causes ACAID (From Jerry Niederkorn’s review in Nature Immunology 7, 354 – 359;2006). Removal of the thymus, eye or spleen within 72 h of injection of antigen into the anterior chamber prevents the induction of ACAID. Chemical sympathectomy before anterior chamber injection of antigen also prevents the induction of ACAID. IL-, interleukin; BCR, B cell receptor.
Several labs have since confirmed that antigens introduced into the AC elicit a deviant immune response, which is characterized by the antigen-specific suppression of classical Th1 immune responses, such as delayed-type hypersensitivity (DTH) and complement-fixing antibodies, while preserving the generation of noncomplement-fixing antibodies of the IgG1 isotype in the mouse. This complex phenomenon called ACAID involves multiple cell types that interact to create unique, antigen-specific immune suppression. Briefly, the injection of antigen into the ocular AC results in the antigen being taken up by circulating F4/80+ cells (a type of dendritic cell) that migrate to the spleen and thymus. Within 3 days of entering the thymus, the F4/80+ cells induce the generation of CD4-CD8-NK1.1+ thymocytes that are believed to enter the circulation as recent thymic emigrants and home to the spleen, where they contribute to the generation of splenic regulatory cells. The spleen is the terminal organ in ACAID, where there is a complex interaction between the F4/80+ cells, natural killer T (NKT) cells, NK1.1 cell, gamma delta T cells and B cells, which in turn elicit the generation of CD4+ and CD8+ regulatory T cells (T regs) specific for the antigen that was introduced in the AC. These T regs are the key players in causing the antigen specific immune-suppression. That ACAID may occur in humans is suggested by the demonstration that individuals with acute retinal necrosis develop antibodies but not cell-mediated immunity to Varicella zoster.
Because the injection of antigen into the AC induces different phenotypes of Tregs and classically Tregs have been known to suppress autoimmunity, Bhowmick et al in 2011 investigated the ability of splenic regulatory T cells induced by an intracameral injection of MOG35-55 peptide to regulate MOG35-55-induced EAE (Experimental Autoimmune Encephalomyelitis), the animal model of human Multiple Sclerosis. In this animal model, MOG35-55 immunization results in an immune response against MOG35-55 peptide which is a component of the Myelin protein, hence causing an inflammatory auto-reaction against myelin and neurodegeneration, similar to the human disease – Multiple Sclerosis. Bhowmick et al found that the injection of MOG35-55 peptide into the ocular AC could suppress MOG35-55 peptide induced EAE, both as a cure (when injected after disease initiation) and as a prophylactic measure (when injected prior to disease induction). The suppression was antigen specific because when they injected an unrelated antigen e.g ovalbumin into the AC, it did not affect EAE in the recipient animal.
They next went on to isolate the AC-injection-induced splenic regulatory T cells and via elegant adoptive transfer experiments showed that AC-induced CD4+ regulatory T cells could suppress the diseases ONLY at the early stage (also known as the priming phase of the disease). While AC-induced CD8+ regulatory T cells could only suppress the progression of an already initiated diseases (known as the chronic phase of EAE). The CD8s were ineffective at the effector phase and vice versa. This was probably the first report which differentiated between the priming and chronic phase of EAE and showed that effective suppression of autoimmune response in EAE can be achieved by different regulatory T cell populations. This work also revealed that while the suppression of EAE by AC-induced CD8+ regulatory T cells is TGF-β dependent, the AC-induced CD4 T regs, do not use TGF-β.
Characteristically, previous studies have shown that ACAID-CD4 Treg cells do not express FoxP3. And the induction of and the activity of regulatory T cells in ACAID is independent of CD4+FoxP3+ regulatory T cells. AC-induced CD8+ regulatory cells suppress IFN-γ production in vitro and in vivo suppress T cells that effect a DTH reaction in immunized mice. Further, AC-induced CD8+ regulatory cells are restricted by Qa-1 antigens expressed by effector T cells. Since the non-classical MHC class I molecule Qa-1 is known to be expressed only on activated cells, AC-induced CD8+ regulatory T cells specifically suppress activated T cells and hence the effector or chronic phase of an autoimmune disease like EAE. Thus it can be concluded that ACAID suppresses the induction of effector T cells and also the activity of effector T cells by distinct populations of regulatory T cells. Recently it was shown that Type II collagen (CII), a key antigen involved in auto-immune responses during Rheumatoid Arthritis, could induce a similar CII-specific immune suppression via ACAID (Farooq et al 2012). A finding that has opened up possibilities of using this system in an Arthritis model to test its efficacy.
Overall these data indicate that the suppression of an ongoing autoimmune disease by the adoptive transfer of regulatory T cells might only be feasible when the regulatory T cells are specific for the pathogenic antigen. Alternatively, transfer of polyclonal regulatory T cells may cure ongoing disease only in lymphopenic hosts, in which the massive expansion of regulatory T cells may lead to the generation of a sufficient number of antigen-specific regulatory T cells. If this is also the case also in humans, it may represent a significant limitation for the clinical application of regulatory T cells in autoimmune diseases, as, to date, human self-antigen specific regulatory T cells have not been successfully expanded ex vivo. In this regard, the use of Anterior Chamber Associated Immune Deviation (ACAID), can be highly effective as ACAID can generate antigen-specific CD8+ and also CD4+ regulatory T cells.

 Arijit Bhowmick is currently a postdoctoral researcher at the Immunology institute of the Mount Sinai Medical Center, NY. He received his PhD in structural immunology from the National Institute of Immunology, New Delhi. His current research interests encompass autoimmunity, Th17 cells and structure based inhibitor designing.
Arijit Bhowmick is currently a postdoctoral researcher at the Immunology institute of the Mount Sinai Medical Center, NY. He received his PhD in structural immunology from the National Institute of Immunology, New Delhi. His current research interests encompass autoimmunity, Th17 cells and structure based inhibitor designing.

