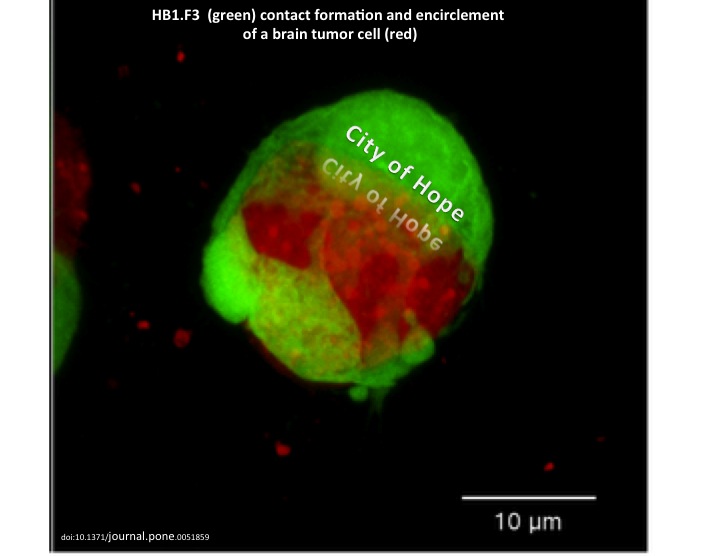
As mentioned in a previous article about Glioblastoma Multiforme (GBM), there are several obstacles that make these lethal brain tumors virtually incurable. Two hallmarks of malignant brain tumors are believed to be the major impediments to effective killing of all tumor cells via the current aggressive course of treatment: glioma cells’ exceptional invasivity and the heterogeneity of GBM, not only among individual patients, but also within a single tumor mass. Thus, these two glioma characteristics have generated great interest in GBM clinical research.

The malignancy of CNS tumors is categorized by the World Health Organization (WHO) grading system, ranging from grade I- grade IV based on the tumor’s “aggressiveness” (GBM is WHO grade IV astrocytoma). However, unlike other neoplasms -in which local dissemination is usually limited and metastasis occurs via the vasculature or the lymphatic system– single glioma cells travel for several centimeters through adjacent brain tissue but will almost never establish systemic metastasis. Thus, the WHO tumor grade positively correlates with proliferation rate, rather than tumor invasion. This suggests the involvement of several independent genetic events that eventually lead to glioma progression, which is also consistent with the high levels of GBM heterogeneity. In order to develop a therapeutic agent that targets (finds and destroys) malignant glioma cells, it’s necessary to find a marker that’s expressed exclusively on the surface of these cells. Due to the high heterogeneity of GBM, as well as variations amnog individual patient brain tumors, no such marker has yet been identified.
There are compelling evidences of a subpopulation of malignant cells in GBM, which exhibit stem-cell like characteristics, such as multipotency, the ability to self-renew and invade; these tumor-initiating cells are referred to as cancer stem cells (CSC) and are believed to be responsible for tumor recurrence in GBM patients. Thus, researchers are no longer focusing on a single mutation/marker. Furthermore, it is becoming prominently important to rely on primary GBM patient samples for collecting data, since glioma cell lines do not represent the cellular and molecular components characteristic of the primary GBM.
CD133 (also known as Prominin-1 or AC133) is a penta-spanning membrane protein with two heavily glycosylated extracellular loops, and is recognized as a stem cell marker for certain normal and cancerous tissues. CD133 accumulates near the Golgi and ER and is also expressed on the cell surface. Although many previous studies reported that the presence of CD133+ glioma CSC subpopulation in GBM, drive tumor formation and its rapid proliferation, subsequent conflicting findings showed CD133-negative glioma cells’ ability to self-renew and form tumors in xeno-transplantation assays.
The recent findings by Brescia’s group confirm that cell-surface CD133 expression is a marker for self-renewing and tumor-initiating GBM cells, but a non-essential element for stem cell properties in all GBM cases. They show that even though membrane-bound CD133 were detectable only in a fraction of the patient samples (neurospheres and in freshly dissociated tumors), CD133 mRNA and intracellular CD133 protein were found expressed at high levels in almost all the examined neurosphere samples.
Through clonal analysis, they demonstrated the intrinsic capabilities of sinlge glioma cells and thier progeny: every clone derived from a single CD133-negative cell, contained a mixture of both CD133-negative and CD133-positive cells. Furthermore, they show an interesting interconvertible regulation of cell-surface CD133, through subcellular localization between the cytoplasm and the plasmamembrane. The localization of CD133 is likely determined by the tumor-associated microenvironmental cues.
The findings in this study have significant value in GBM research, taking one step closer to understanding the progression of malignant gliomas. However, the existence of the cytoplasmic CD133 reservoir and the re-cycling of this protein to the plasmamembrane and vise versa, hinders the therapeutic potential of using CD133 as a glioblastoma-targeting marker.

In a separate study published in Neuro-Oncology on the same week, researchers at Mayo Clinic introduced a significant association between malignant gliomas and Kallikrein 6 (KLK6) enzyme. KLK6 is a member of the kallikrein family of secreted serine proteases and has been reported to be elevated within areas of inflammation in CNS, suggesting its regulated expression with T-cell activation. Notably, the serum of Multiple Sclerosis (MS) patients contains elevated KLK6 levels as well. This study shows that as the KLK6 expression levels increase, the post-surgery survival times of GBM patients decrease. They found the highest levels of KLK6 were present in the most severe GBM. These results were obtained by looking at 60 samples of grade IV astrocytomas (thus categorized as GBM), and a less aggressive grade III astrocytomas.
Scarisbrick’s group also showed the possible role of KLK6 in promoting survival of malignant glioma cells, as well as increasing their resistance to apoptosis-inducing agents, such as radiotherapy (RT) and temozolomide (TMZ). The data supporting the pro-survival effect of KLK6 may introduce a new GBM therapeutic strategy, albeit the supporting experiments of these observations were conducted on U251 glioma cell line. If future studies report similar results that confirm the ability of KLK6 enzyme to promote primary patient tumor cells’ survival, then developing a therapeutic agent which targets KLK6’s function is a promising addition to GBM course of treatment ensuing the surgical resection of the tumor, preceding chemo-and radiotherapy.
Further Reading:



 96-well Plates: Different types of 96-well plates are available for different assay types. There are various surface coatings including tissue-culture treated polystyrene for cell cultures, uncoated, and others. Plates can have various plate bottom geometries and optical characteristics. For instance there are black plates available for light-sensitive assays. For protocols involving volumes larger then 250ul, there are deep-well plates that carry a 2ml volume per well.
96-well Plates: Different types of 96-well plates are available for different assay types. There are various surface coatings including tissue-culture treated polystyrene for cell cultures, uncoated, and others. Plates can have various plate bottom geometries and optical characteristics. For instance there are black plates available for light-sensitive assays. For protocols involving volumes larger then 250ul, there are deep-well plates that carry a 2ml volume per well. Multichannel Vacuums: Companies such as V&P Scientific offer a multitude of multichannel vacuum manifolds that fit plates of different depths for removing supernatant from wells via vacuum apparatus. Often these will be the proper length such that they don’t touch the well bottom and work well with removing buffers from centrifuged PBMC cell cultures, such as during washing steps for flow-cytometry.
Multichannel Vacuums: Companies such as V&P Scientific offer a multitude of multichannel vacuum manifolds that fit plates of different depths for removing supernatant from wells via vacuum apparatus. Often these will be the proper length such that they don’t touch the well bottom and work well with removing buffers from centrifuged PBMC cell cultures, such as during washing steps for flow-cytometry.