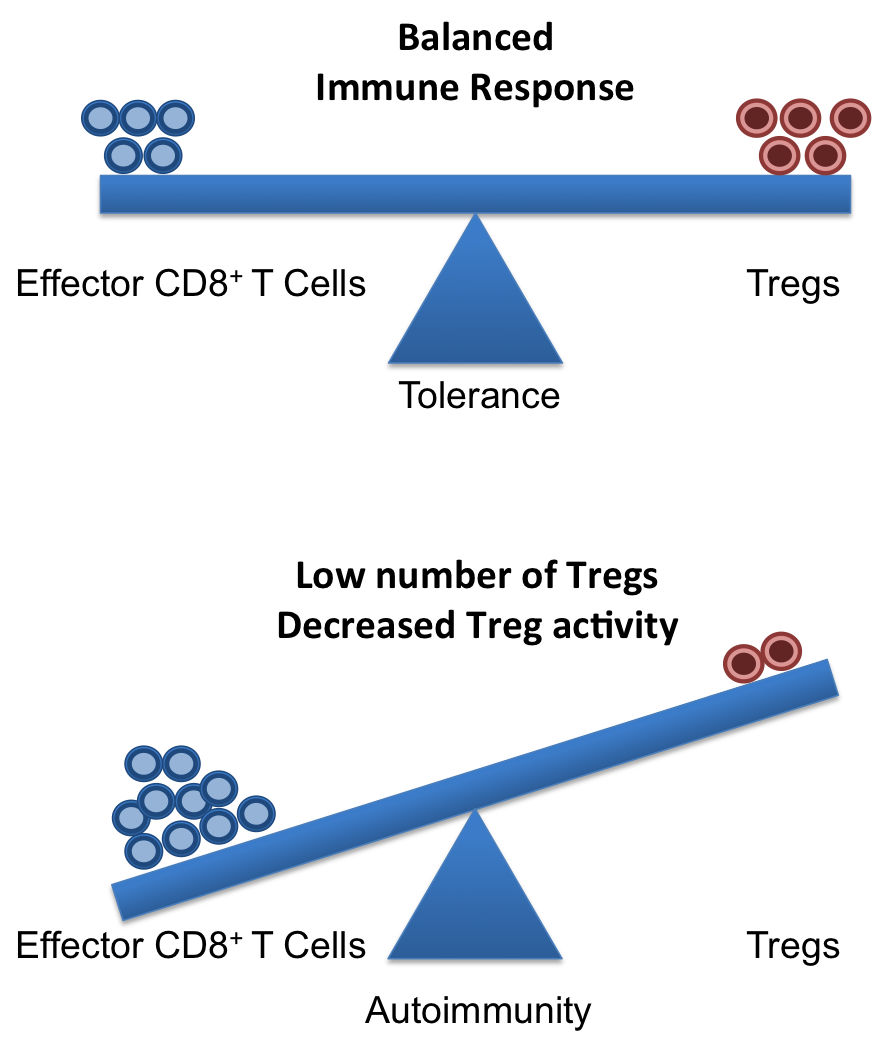
A healthy and functional immune system requires a delicate balance of pro- and contra-inflammatory signals. Whereas, it is important to induce a strong and efficient immune response against pathogens, it is similarly important to dampen these responses after the pathogen is fought off to revert the immune system to a calm steady state. If the balance is disturbed, diseases can on the one hand, become chronic/overwhelming or, on the other hand, inflammatory responses that cannot terminate can result in autoimmune responses.
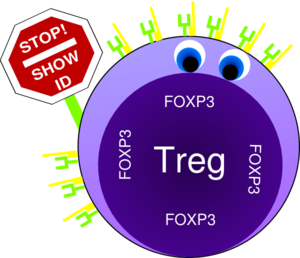
Crucial elements in the regulation of excessive immune responses are regulatory T (Treg) cells. Tregs are known to inhibit the response of other immune cells. Their essential role in limiting overwhelming immune responses is demonstrated by the detrimental consequences of their loss. Mice or humans lacking Tregs develop widespread and lethal autoimmune diseases. Besides several surface markers, Tregs are best characterized by the expression of the transcription factor FoxP3. This factor is essential for Treg function and its artificial expression in other T cells can induce a regulatory potential. Therefore, the expression of FoxP3 is required for a T cell to have regulatory potential (Buckner; Josefowicz et al.). However, it was known for many years that in cases of numerous autoimmune diseases FoxP3+ Tregs could be found in high numbers at the sides of inflammation, but that they did not demonstrate any or not sufficient regulatory activity. This enigmatic observation was so far poorly understood (Buckner; Josefowicz et al.).
In the March 2013 issue of Nature Medicine Nie and colleagues shed new light on the underlying mechanism that impairs Treg function at the sites of inflammation. Studying Treg cells from rheumatoid arthritis (RA) patients the authors demonstrated that phosphorylation of FoxP3 of the serine at position 418 (S418) is required for its regulatory action. If FoxP3 lacks this particular phosphorylation the Treg cell is not suppressive! FoxP3 S418 in Tregs is usually phosphorylated and hence Tregs are regulatory by default. However, the authors show that due to the action of the enzyme ‘protein phosphatase 1’ (PP1) FoxP3 can lose its S418 phosphorylation. Intriguingly, the presence of the cytokine TNF lead to an up-regulation of PP1 expression in the Tregs in a dose-dependent manner, and this lead to de-phosphorylation of FoxP3 S418. Treg cells expressing a mutant FoxP3 that replaced the serine at position 418 with an alanine retained their suppressive potential even in the presence of TNF, demonstrating the importance of the phosphorylation of S418. With this finding, the authors were able to link the pro-inflammatory milieu (TNF) to a specific effect inside of the Tregs (de-phosphorylation of S418) that lead to the observed loss of the regulatory function of Treg cells. Importantly, the authors were also able to demonstrate the therapeutic potential of this knowledge. They monitored RA patients that underwent treatment with blocking anti-TNF antibodies (infliximab) and found that Tregs from patient PBMCs restored S418 phosphorylation and regained regulatory potential!
This is the second case for a post-transcriptional regulation of FoxP3 that can influence Treg function. Deacetylation of FoxP3 has been linked to impaired Treg function previously (Tao et al.). Additionally, the work of Nie et al. now adds mechanistic information to previous reports on the negative effect of TNF on Tregs (Valencia et al.; Zanin-Zhorov et al.).
Given the ubiquitous role of TNF during inflammation, it is very likely that the mechanism described by Nie et al. applies to many if not all cases of ongoing inflammation where Treg function is impaired. Furthermore, their data on the effects of anti-TNF antibody treatment in RA suggest a similar therapeutic potential in other autoimmune diseases. Surely, this report will ignite further investigation in this direction and will aid the development of better treatments for patients suffering from autoimmune diseases.
References:
Bromberg, J., 2013. TNF-α trips up Treg cells in rheumatoid arthritis. Nat Med, 19(3), pp.269–270.
Buckner, J.H., 2010. Mechanisms of impaired regulation by CD4(+)CD25(+)FOXP3(+) regulatory T cells in human autoimmune diseases. Nat Rev Immunol, 10(12), pp.849–859.
Josefowicz, S.Z., Lu, L.-F. & Rudensky, A.Y., 2012. Regulatory T cells: mechanisms of differentiation and function. Annual Review of Immunology, 30, pp.531–564.
Nie, H. et al., 2013. Phosphorylation of FOXP3 controls regulatory T cell function and is inhibited by TNF-α in rheumatoid arthritis. Nat Med, 19(3), pp.322–328.
Tao, R. et al., 2007. Deacetylase inhibition promotes the generation and function of regulatory T cells. Nature Medicine, 13(11), pp.1299–1307.
Valencia, X. et al., 2006. TNF downmodulates the function of human CD4+CD25hi T-regulatory cells. Blood, 108(1), pp.253–261.
Zanin-Zhorov, A. et al., 2010. Protein kinase C-theta mediates negative feedback on regulatory T cell function. Science, 328(5976), pp.372–376.