
Breast cancer is the most common cancer in women and the second-leading cause of cancer-related death in women worldwide. Despite progresses in the treatment of early stage breast cancer, approximately one third of patients will develop metastatic breast cancer (MBC). According to the National Cancer Institute, in USA, the estimated new cases and deaths from breast cancer in 2013 would be 232,340 and 39,620 respectively.
Approximately 20%–30% of breast cancers exhibit increased expression of human epidermal growth factor receptor 2 (HER-2/neu) caused by amplification of the erb-B2 oncogene. Breast cancers with elevated HER-2 expression are known as HER2-positive cancers. HER-2-positive breast cancers are more aggressive than other breast cancers. Patients with these tumors have a poorer prognosis and decreased chance of survival compared with patients whose tumors do not overexpress HER-2.
 HER-2 is a 185-kDa orphan transmembrane receptor tyrosine kinase. Dimerization of HER-2 with ligand- bound HER-3 or HER-4 receptor activates signaling pathways inside the cell. Activated HER-2 signaling stimulates cell proliferation and survival via activation of the MAPK and PI3K/Akt/mTOR pathways. Collectively these signaling pathways result in uncontrolled growth of the tumor. Several studies suggested that the overexpression/amplification of HER-2 may lead to the development and progression of pre-malignant breast disease and also tumor metastasis. Therefore, the association of HER-2 in breast cancer as well as its involvement in tumor aggressiveness makes this receptor an appropriate target for tumor-specific therapies. Several strategies have been developed to inhibit HER-2 signaling. These include a tyrosine kinase inhibitor called lapatinib and a recombinant humanized monoclonal antibody called trastuzumab (Herceptin®). In this post I will focus only on trastuzumab mediated therapy in breast cancer. Trastuzumab binds to the extracellular domain of the HER-2 receptor. This inhibits HER-2 signaling via MAPK and PI3K/Akt cascades. In addition, trastuzumab binding also increases membrane localization of the tumor suppressor gene phosphatase and tensin homolog (PTEN), and inhibitor of the PI3K/Aktpathway.
HER-2 is a 185-kDa orphan transmembrane receptor tyrosine kinase. Dimerization of HER-2 with ligand- bound HER-3 or HER-4 receptor activates signaling pathways inside the cell. Activated HER-2 signaling stimulates cell proliferation and survival via activation of the MAPK and PI3K/Akt/mTOR pathways. Collectively these signaling pathways result in uncontrolled growth of the tumor. Several studies suggested that the overexpression/amplification of HER-2 may lead to the development and progression of pre-malignant breast disease and also tumor metastasis. Therefore, the association of HER-2 in breast cancer as well as its involvement in tumor aggressiveness makes this receptor an appropriate target for tumor-specific therapies. Several strategies have been developed to inhibit HER-2 signaling. These include a tyrosine kinase inhibitor called lapatinib and a recombinant humanized monoclonal antibody called trastuzumab (Herceptin®). In this post I will focus only on trastuzumab mediated therapy in breast cancer. Trastuzumab binds to the extracellular domain of the HER-2 receptor. This inhibits HER-2 signaling via MAPK and PI3K/Akt cascades. In addition, trastuzumab binding also increases membrane localization of the tumor suppressor gene phosphatase and tensin homolog (PTEN), and inhibitor of the PI3K/Aktpathway.
In 1998 trastuzumab was approved for thetreatment of metastatic breast cancer (MBC), and in 2006 for the adjuvant treatment of HER2-overexpressing breast cancer. In early-stage breast cancer, treatment with trastuzumab and a neoadjuvant chemotherapy substantially improves overall survival (OS) and reduces the risk of recurrence, both by 33%. In MBC, trastuzumab treatment in combination with chemotherapy increases the time to progression of the disease by 49% and improves OS by 20%.
However, even though trastuzumab treatment substantially improves outcomes in both early-stage and MBC, both de novo and acquired resistance after initial response was observed. It is suggested that most patients with HER2-positive MBC will eventually develop resistance and have disease progression following trastuzumab treatment.
Several studies reported involvement of multiple factors in resistance to HER2-targeted therapy. These include hindrance to HER-2-trastuzumab binding, signaling through alternative pathways (for e.g. insulin-like growth factor receptor 1, vascular endothelial growth factor receptor) upregulation of signaling pathways downstream of HER-2, increased expression of heat shock protein 90 (HSP90), loss of PTEN and thereby constitutive activation of the PI3K/Akt pathway, and failure to induce an appropriate immune response.
To overcome transtuzumab-resistance, various treatment strategies have been developed. One strategy involves continuation of transtuzumab treatment in combination with a chemotherapeutic agent. In multiple pre-clinical and clinical studies, combination of  trastuzumab with taxanes docetaxel (Taxotere®) and paclitaxel (Taxol®) exhibited promising response in HER-2–overexpressing metastatic breast cancer.
trastuzumab with taxanes docetaxel (Taxotere®) and paclitaxel (Taxol®) exhibited promising response in HER-2–overexpressing metastatic breast cancer.
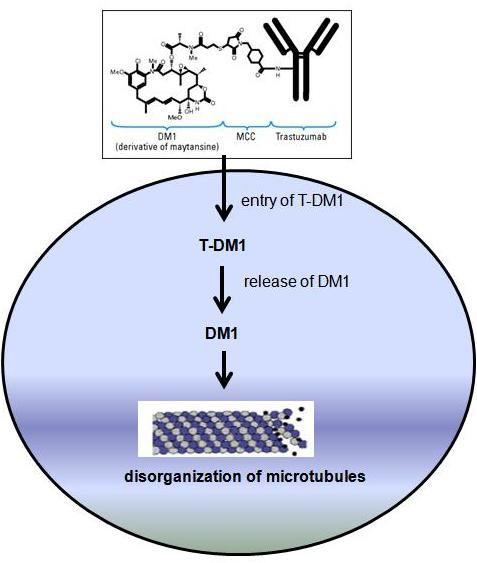
A new strategy to increase efficacy of trastuzumab has also been developed using antibody-drug conjugate (ADC) technology. The antibody-drug conjugate trantuzumab emtansine (T-DM1, Kadcyla) is consist of trastuzumab bound to maytansinoid (or DM1, a potent microtubule inhibitor) through a nonreducible thioether linkage. T-DM1 binds to HER-2 positive tumor cells and thought to inhibit HER-2 signaling. This ADC also induces body’s immune response to attack cancer cells. Once inside the tumor cells, T-DM1 is designed to kill tumor cells by releasing DM1 which is a potent inhibitor of microtubule assembly, thereby causing cell death inside the cells.
In in vitro and preclinical studies T-DM1 inhibited growth of breast cancer cells which are cross-resistant to trastuzumab. T-DM1 was found well tolerated in phase I clinical study of breast cancer patients who had disease progression with earlier trastuzumab based treatment. In phase II study, increased progression-free survival (PFS) was observed in patients treated with T-DM1 compared to trastuzumab plus doecetaxel treatment. A clinical study published by Verma et al. (2012) reported that T-DM1 significantly prolonged PFS and OS in patients with HER-2 positive MBC previously treated with trastuzumab and a taxane. The most common side effects of T-DM1 treatment include low platelet count, low RBC count, nerve problems, and tiredness. On the basis of clinical efficacy of T-DM1 observed in phase I and II trials, a multicenter phase III trial (also known as EMILIA trial) was performed. This trial also observed increased PFS, reduction of risk of death, and fewer adverse events in T-DM1 treated patients compared to capecitabine plus lapatinib treatment (another first-line treatment option for HER-2positive MBC).
On February 22nd, 2013, the US food and drug administration (FDA) approved T-DM1 (Kadcyla) for the treatment of HER-2 positive MBC that has progressed following treatment with trastuzumab and a taxane.
Suggested reading:
[1] M.F. Barginear, V. John, D.R. Budman, Trastuzumab-DM1: A Clinical Update of the Novel Antibody-Drug Conjugate for HER2-Overexpressing Breast Cancer, Mol Med, 18 (2013) 1473-1479.
[2] M. Barok, M. Tanner, K. Köninki, J. Isola, Trastuzumab-DM1 causes tumour growth inhibition by mitotic catastrophe in trastuzumab-resistant breast cancer cells in vivo, Breast Cancer Res, 13 (2011) R46.
[3] M.S. Mohd Sharial, J. Crown, B.T. Hennessy, Overcoming resistance and restoring sensitivity to HER2-targeted therapies in breast cancer, Ann Oncol, 23 (2012) 3007-3016.
[4] S. Verma, D. Miles, L. Gianni, I.E. Krop, M. Welslau, J. Baselga, M. Pegram, D.Y. Oh, V. Diéras, E. Guardino, L. Fang, M.W. Lu, S. Olsen, K. Blackwell, E.S. Group, Trastuzumab emtansine for HER2-positive advanced breast cancer, N Engl J Med, 367 (2012) 1783-1791.
[5]http://www.cancer.gov/cancertopics/understandingcancer/targetedtherapies/breastcancer_htmlcourse/page3
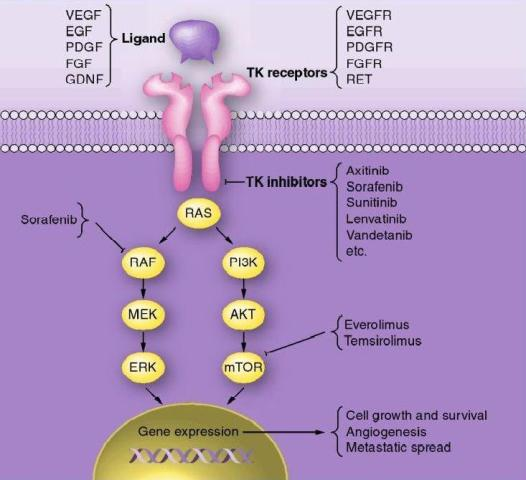
 RAS and BRAF oncogenes. In follicular thyroid carcinoma mutations of RAS oncogene in addition to other gene mutations was also observed. Aberrant activities of these oncogenes results in constitutive activation of the MAPK-pathway leading to inhibition of sodium-iodide symporter and thyroid peroxidase genes which participate in iodide uptake and thyroid hormone production respectively. In a pre-clinical study, Chakravarty et al. (2011) showed that mice with poorly differentiating thyroid cancer and overexpressing BRAF (V600E) oncogene failed to uptake RAI. Shutting BRAF activation off or inhibiting the MAPK-pathway with kinase inhibitors targeting BRAF or MEK rendered mice susceptible to a therapeutic dose of RAI. This suggests that inhibition of the MAPK-pathway and its associated protein kinases with the protein kinase inhibitors may facilitate RAI uptake in refractory thyroid cancer patients harboring MAPK-pathway activation.
RAS and BRAF oncogenes. In follicular thyroid carcinoma mutations of RAS oncogene in addition to other gene mutations was also observed. Aberrant activities of these oncogenes results in constitutive activation of the MAPK-pathway leading to inhibition of sodium-iodide symporter and thyroid peroxidase genes which participate in iodide uptake and thyroid hormone production respectively. In a pre-clinical study, Chakravarty et al. (2011) showed that mice with poorly differentiating thyroid cancer and overexpressing BRAF (V600E) oncogene failed to uptake RAI. Shutting BRAF activation off or inhibiting the MAPK-pathway with kinase inhibitors targeting BRAF or MEK rendered mice susceptible to a therapeutic dose of RAI. This suggests that inhibition of the MAPK-pathway and its associated protein kinases with the protein kinase inhibitors may facilitate RAI uptake in refractory thyroid cancer patients harboring MAPK-pathway activation. resistant to RAI. Selumetinib is currently in clinical trials for various solid and hematologic malignancies. Among 24 patients screened for the study 5 had NRAS-mutant tumors. All of them showed augmented RAI uptake following treatment with selumetinib; 4 patients exhibited confirmed partial responses (PR) and in 1 patient no disease progression was noted following RAI treatment. No significant levels of toxic effects to selumetinib were observed in this study. Increased RAI uptake and confirmed PR was also observed in 1 patient with BRAF mutation after treatment with selumetinib. However, selumetinib treatment in majority of the patients with BRAF mutations enrolled in this study did not increase RAI uptake up to the threshold level required for therapy. Therefore, further studies are required to understand the differences observed between RAS-mutant and BRAF-mutant tumors.
resistant to RAI. Selumetinib is currently in clinical trials for various solid and hematologic malignancies. Among 24 patients screened for the study 5 had NRAS-mutant tumors. All of them showed augmented RAI uptake following treatment with selumetinib; 4 patients exhibited confirmed partial responses (PR) and in 1 patient no disease progression was noted following RAI treatment. No significant levels of toxic effects to selumetinib were observed in this study. Increased RAI uptake and confirmed PR was also observed in 1 patient with BRAF mutation after treatment with selumetinib. However, selumetinib treatment in majority of the patients with BRAF mutations enrolled in this study did not increase RAI uptake up to the threshold level required for therapy. Therefore, further studies are required to understand the differences observed between RAS-mutant and BRAF-mutant tumors.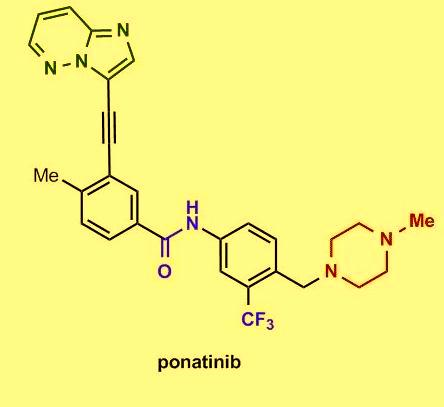
 Several in vitro studies showed that the tyrosine kinase chimeric protein Bcr-Abl encoded by the BCR-ABL gene is constitutively active in leukemia cells and has oncogenic properties. Bcr-Abl chimeric protein has been found to be associated with genomic instability and thereby suggested to be responsible for progression to advanced phases of CML.
Several in vitro studies showed that the tyrosine kinase chimeric protein Bcr-Abl encoded by the BCR-ABL gene is constitutively active in leukemia cells and has oncogenic properties. Bcr-Abl chimeric protein has been found to be associated with genomic instability and thereby suggested to be responsible for progression to advanced phases of CML. Among chronic-phase CML patients with T315I mutation 100% exhibited hematologic response, 92% had a major cytogenetic response, 75% exhibited complete cytogenetic response, and 67% had a major molecular response. The most common side effects reported in the study include hypertension, rash, abdominal pain, fatigue, headache, dry skin, constipation, fever, joint pain, and nausea. Clinically promising similar results were also observed in the PACE trial, a multicenter, international, single-arm clinical trial of 449 patients with disease that was resistant or intolerant to prior tyrosine kinase inhibitor therapy. On December 14, 2012, the FDA approved ponatinib (Iclusig tablets) for the treatment of adult patients with all phases of CML that are resistant or intolerant to prior tyrosine kinase inhibitor therapy.
Among chronic-phase CML patients with T315I mutation 100% exhibited hematologic response, 92% had a major cytogenetic response, 75% exhibited complete cytogenetic response, and 67% had a major molecular response. The most common side effects reported in the study include hypertension, rash, abdominal pain, fatigue, headache, dry skin, constipation, fever, joint pain, and nausea. Clinically promising similar results were also observed in the PACE trial, a multicenter, international, single-arm clinical trial of 449 patients with disease that was resistant or intolerant to prior tyrosine kinase inhibitor therapy. On December 14, 2012, the FDA approved ponatinib (Iclusig tablets) for the treatment of adult patients with all phases of CML that are resistant or intolerant to prior tyrosine kinase inhibitor therapy.
 R4) and TR2 (DR5). TRAIL also binds with two other receptors TR3 and TR4. Unlike TR1 and TR2, these receptors have incomplete death domains. Elevated expression of TR3 and TR4 in normal cells is thereby suggested to protect normal cells from TRAIL-induced death signaling. Induction of apoptotic signaling involves binding of TRAIL to DR4 or DR5. This results in homotrimerization and activation of receptors, enabling the receptors’ death domain to recruit the adaptor protein Fas-associated death domain along with pro-caspase- 8 and pro-caspase- 10. These all together then form the multi-protein death-inducing signaling complex, (DISC). Inside the DISC procaspeses become autoactivated and become caspase-8/10. Activated caspase-8/10 then activates downstream effector caspase-3 or caspase-7. Finally, cleavage of downstream substrates by effector caspases results in DNA fragmentation leading to apoptosis.
R4) and TR2 (DR5). TRAIL also binds with two other receptors TR3 and TR4. Unlike TR1 and TR2, these receptors have incomplete death domains. Elevated expression of TR3 and TR4 in normal cells is thereby suggested to protect normal cells from TRAIL-induced death signaling. Induction of apoptotic signaling involves binding of TRAIL to DR4 or DR5. This results in homotrimerization and activation of receptors, enabling the receptors’ death domain to recruit the adaptor protein Fas-associated death domain along with pro-caspase- 8 and pro-caspase- 10. These all together then form the multi-protein death-inducing signaling complex, (DISC). Inside the DISC procaspeses become autoactivated and become caspase-8/10. Activated caspase-8/10 then activates downstream effector caspase-3 or caspase-7. Finally, cleavage of downstream substrates by effector caspases results in DNA fragmentation leading to apoptosis. gen-activated protein kinase (MAPK) pathways was noted following TIC10 treatment in this in vivo study that resulted in translocation of the transcription factor Foxo3a into the nucleus, where Foxo3a induced expression of TRAIL gene to activate apoptosis.
gen-activated protein kinase (MAPK) pathways was noted following TIC10 treatment in this in vivo study that resulted in translocation of the transcription factor Foxo3a into the nucleus, where Foxo3a induced expression of TRAIL gene to activate apoptosis.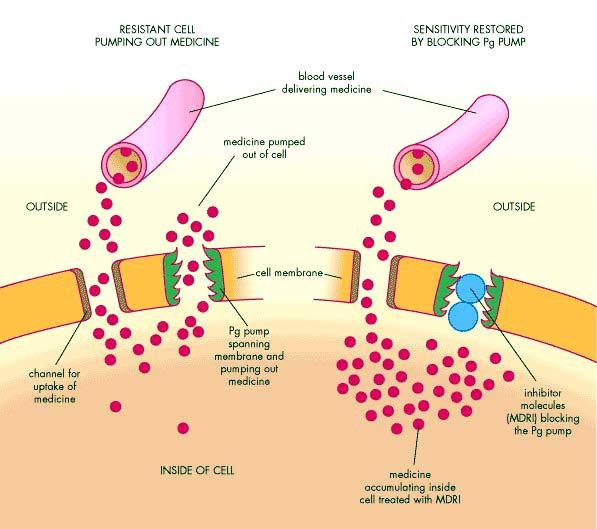
 As shown in Figure 1, tumor cells adopt several mechanisms to evade death induced by anti-tumor agents. These include changes in apoptotic pathways and activation of cell-cycle check points to increase DNA repair. Alternatively, cancer cells develop resistance by increased expression of multidrug-resistant proteins and altered anti-tumor drug transport mechanisms. Members of the ABC transporters (ATP-binding cassette) are known to be associated with this phenomenon, as the human genome express over 48 genes in this transporter family alone. These proteins bind ATP in their ATP binding domain and use the energy to transport various molecules across the cell, thus they are known as ABC proteins. Among these proteins, P-glycoprotein (Pgp, ABCB1), multidrug resistance-associated protein (MRP1, ABCC1), and breast cancer resistance protein (BCRP, ABCG2) are chiefly responsible for drug resistance in tumor cells. Studies are warranted to determine the role of other members of ABC transporters including MRP2, MRP3, MRP4, MRP5, ABCA2 and BSEP in drug-resistance.
As shown in Figure 1, tumor cells adopt several mechanisms to evade death induced by anti-tumor agents. These include changes in apoptotic pathways and activation of cell-cycle check points to increase DNA repair. Alternatively, cancer cells develop resistance by increased expression of multidrug-resistant proteins and altered anti-tumor drug transport mechanisms. Members of the ABC transporters (ATP-binding cassette) are known to be associated with this phenomenon, as the human genome express over 48 genes in this transporter family alone. These proteins bind ATP in their ATP binding domain and use the energy to transport various molecules across the cell, thus they are known as ABC proteins. Among these proteins, P-glycoprotein (Pgp, ABCB1), multidrug resistance-associated protein (MRP1, ABCC1), and breast cancer resistance protein (BCRP, ABCG2) are chiefly responsible for drug resistance in tumor cells. Studies are warranted to determine the role of other members of ABC transporters including MRP2, MRP3, MRP4, MRP5, ABCA2 and BSEP in drug-resistance. Plasma membrane glycoprotein (Pgp) was the first ABC-transporter detected in various cancers exerting resistance to a variety of chemically unrelated cytotoxic agents including anti-tumor drugs such as doxorubicin, vinblastine, ritonavir, indinavir and paclitaxel. It works as an energy-dependent efflux pump and can recognize a wide range of substrates. Even though this protein normally protects us from endogenous and exogenous toxins by transporting them out of the cells, the transporter causes a major problem in the bioavailability of anti-tumor drugs to tumor cells during chemotherapy.
Plasma membrane glycoprotein (Pgp) was the first ABC-transporter detected in various cancers exerting resistance to a variety of chemically unrelated cytotoxic agents including anti-tumor drugs such as doxorubicin, vinblastine, ritonavir, indinavir and paclitaxel. It works as an energy-dependent efflux pump and can recognize a wide range of substrates. Even though this protein normally protects us from endogenous and exogenous toxins by transporting them out of the cells, the transporter causes a major problem in the bioavailability of anti-tumor drugs to tumor cells during chemotherapy.
 histone tails. HDACs also cause deacetylation of non-histone proteins thus altering the transcriptional activity of p53 (tumor suppressor gene), E2F (transcription factor), c-Myc (transcription factor), nuclear factor kB (NF-kB), hypoxia inducible factor 1α (HIF-1 α), estrogen receptor α, and androgen receptor complexes.
histone tails. HDACs also cause deacetylation of non-histone proteins thus altering the transcriptional activity of p53 (tumor suppressor gene), E2F (transcription factor), c-Myc (transcription factor), nuclear factor kB (NF-kB), hypoxia inducible factor 1α (HIF-1 α), estrogen receptor α, and androgen receptor complexes. currently in clinical trials both in monotherapy and in combination therapy with other anti-tumor drugs. A review by Tan et al. (2010) reported that at least 80 clinical trials are underway, testing more than 11 different HDIs in hematologic and solid tumors, including leukemias, lymphomas, and multiple myeloma, lung, breast, pancreas, renal, and bladder cancers, melanoma, glioblastoma. To date, most of the responses using HDIs as single agents were observed in advanced hematologic tumors and few were observed in solid tumors. In 2006, HDI vorinostat (suberoylanilide hydroxamic acid, SAHA) was approved by the Food and Drug Administration (FDA, USA) for the treatment of relapsed and refractory cutaneous T-cell lymphoma CTCL. In November, 2009, the FDA also approved another HDI romidepsin (depsipeptide) for the treatment of CTCL, and in 2011 for the treatment of peripheral T-cell lymphoma patients who have already received prior therapy.
currently in clinical trials both in monotherapy and in combination therapy with other anti-tumor drugs. A review by Tan et al. (2010) reported that at least 80 clinical trials are underway, testing more than 11 different HDIs in hematologic and solid tumors, including leukemias, lymphomas, and multiple myeloma, lung, breast, pancreas, renal, and bladder cancers, melanoma, glioblastoma. To date, most of the responses using HDIs as single agents were observed in advanced hematologic tumors and few were observed in solid tumors. In 2006, HDI vorinostat (suberoylanilide hydroxamic acid, SAHA) was approved by the Food and Drug Administration (FDA, USA) for the treatment of relapsed and refractory cutaneous T-cell lymphoma CTCL. In November, 2009, the FDA also approved another HDI romidepsin (depsipeptide) for the treatment of CTCL, and in 2011 for the treatment of peripheral T-cell lymphoma patients who have already received prior therapy.