
Shinya Yamanaka was awarded the Noble Prize for Medicine last year for his work on cellular reprogramming and creating induced pluripotent stem cells (iPSC). Shinya Yamanaka found four transcription factors (Oct-3/4, Sox2, c-Myc, and Klf4) that determine pluripotency and he was able to reprogram differentiated adult cells into pluripotent cells that can then be re-differentiated into fully specialized tissue. His findings raised great expectations, especially in the field of cellular therapy and regenerative medicine. However, the path to the therapeutic use of iPSC is long and not without complications. It was believed that iPSCs would avoid any immunogenic response because these cells can be developed from a patient’s own somatic cells. However, Zhao et al. challenged this notion when they discovered that iPSCs derived from C57BL/6 (B6) mice by the standard retroviral approach formed teratomas once transplanted into syngenic host mice and induced a rapid T cell dependent immune response1.
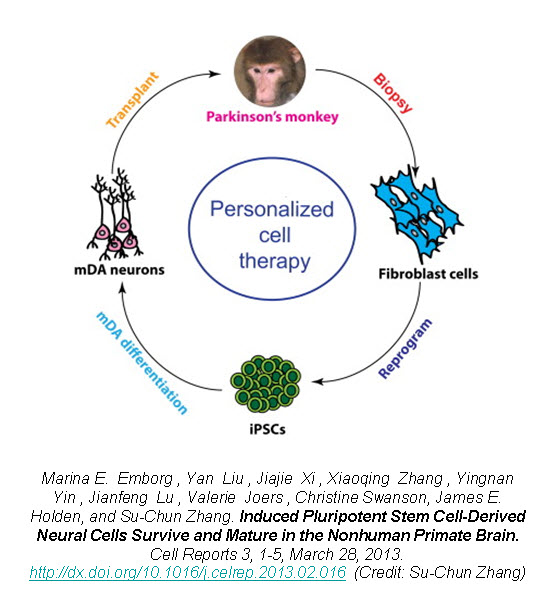
Recently, a group of researchers led by Dr. Su-Chun Zhang at the Waisman Center on the University of Wisconsin-Madison campus have shown that utilizing iPSCs as an autologous cell therapy is feasible and without any immunological reaction or rejection. Dr. Zhang was the first to derive neural cells from embryonic stem cells (ESCs), as well as from iPSCs, and now has shown the first proof-of-principle that autologous iPSC-derived cells can engraft and survive in the primate brain.
In a new study published in Cell Reports, Zhang’s group reports on the successful generation of iPSCs from fibroblasts obtained from 8-10 year old rhesus monkeys (Macaca mulatta) using retroviruses containing the four Yamanaka factors, subsequent neuronal differentiation and cell transplantation back into the donor monkeys2. The rhesus iPSCs were differentiated into neuroepithelia with the characteristic neural tube-like rosettes and expression of neuroectoderm transcription factors Pax6 and Sox1. The neuronal rosettes were then expanded and further differentiated into neurons so that by the time for cell transplantation (day 42), 37% of the cells were bIII-tubulin+ neurons, 16% were S100b+ immature astrocytes, and 47% were Nestin+ progenitors.

To examine the feasibility of transplanting autologous iPSC-derived neural progenitors in Parkinson’s disease, Zhang created parkinsonism in the rhesus monkeys by unilateral intracarotid artery injection of a neurotoxin 1-methyl-4-phenyl-1,2,3,6,-tetrahydropyridine (MPTP). A year to 18 months after the MPTP infusion, all the monkeys developed a stable hemiparkinsonian condition distinguished by the characteristic tremors, bradykinesia, imbalance and impairment in motor skills. At day 30 of cell culture the iPSC-derived neural progenitors were labeled with a GFP (green fluorescent protein) lentivirus and cells were transplanted at day 42 into the striatum and substantia nigra of the same monkey from which the iPSC were derived.
The monkey did not receive any immune suppression and engraftment of the GFP-labeled iPSC-derived neural progenitors was assessed at 6 months post-transplantation using stereological analysis on serial coronal sections. Distinct grafts were present in the injected regions where 63% of the cells were microtubule-associated protein 2 (MAP2)+ neurons, 22% were glial fibrillary acidic protein (GFAP)+ astrocytes, and 10% were myelin basic protein (MBP)+ oligodendrocytes. The GFP+ neurons showed long fibers extending into the surrounding host tissue and some neurons were present outside of the graft region expressing markers of mature neuronal differentiation. Additionally, there was an absence of markers for pluripotent stem cells (OCT4, NANOG, SOX17, and Brachyury) and no positive staining for Ki67 (labels mitotic cells) indicating that the grafted progenitors had terminally differentiated.
Zhao et al. showed that the teratomas formed from transplanted iPSCs had an immunological rejection by syngenic mice1. However, Zhang demonstrates that the autologous iPSC-derived neural cell transplants in the primate brain do not undergo immunological rejections suggested by the lack of CD3 and CD8 (lymphocyte markers) staining. There was minimal response of endogenous glia (astrocytes and microglia) since staining for human leukocyte antigen D-related (HLA-DR; microglia and macrophage marker) was seen throughout the brain including the grafted regions.
Despite the positive engraftment and differentiation of the iPSC-derived neural progenitors, they did not see any behavioral improvement in the parkinsonian monkeys. A potential explanation is that the GFP+ neurons were mostly g-Aminobutyric acid (GABA+) and few were positive for tyrosine hydroxylase (TH+). TH catalyzes the formation of L-DOPA, the rate-limiting step in the biosynthesis of dopamine. Deficiency in TH has been implicated in giving rise to parkinsonian characteristics3. Also, the number of transplanted cells may not have been enough to replace the dopamine-making cells in the primate brain.
However, this study provides hope for cell therapy using autologous iPSC-derived cells. The iPSC-derived neural progenitors survived and differentiated into mature neurons, astrocytes, and oligodendrocytes in the primate brain with no evidence of immune rejection or teratoma formation. The transplanted cells structurally integrated into the host brain and with characteristic features of neurons indicating by extending long processes and features of oligodendrocytes indicated by staining for myelin basic protein, suggestive of myelination. This proof-of-principle study of the first-ever transplant of iPSC-derived cells back into the same non-human primate presents hope for personalized regenerative medicine and the neurodegenerative patient population.
Further reading:
1. Zhao, Tongbiao; Zhang, Zhen-Ning; Rong, Zhili; and Xu, Yang. Immunogenicity of induced pluripotent stem cells. Nature. 474, 212–215, 9 June 2011
2. Emborg ME, Liu Y, Xi J, Chang X, Yin Y, Lu J, Joers V, Swanson C, Holden JE, and Zhang Su. Induced pluripotent stem cell-derived neural stem cells survive and mature in the nonhuman primate brain. Cell Reports 3, 1-5, March 28, 2013.
3. Goodwill KE, Sabatier C, Marks C, Raag R, Fitzpatrick PF, Stevens RC. Crystal Structure of tyrosine hydroxylase at 2.3a and its implications for inherited neurodegenerative diseases. Nature Structural Biology 4 (7): 578–585, 1997
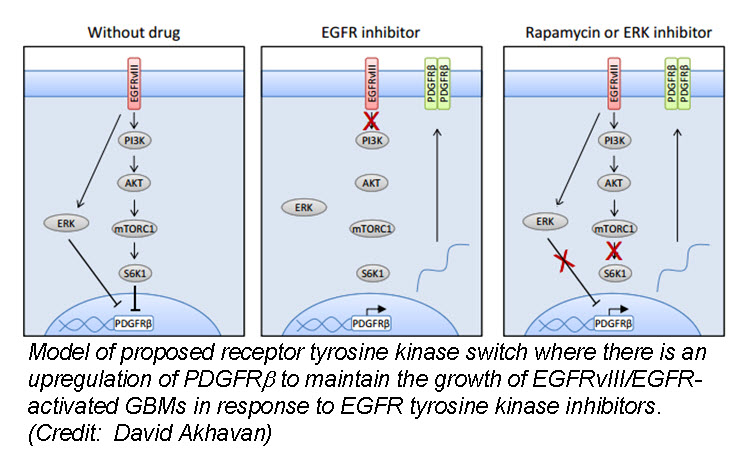
 a (GBM) cells develop resistance to anti-EGFR therapy. They demonstrate for the first time that an EGFR-dependent cancer can escape targeted therapy by developing dependence on another non-amplified, non-mutated RTK. Specifically, they show that GBMs with EGFR mutations evade EGFR tyrosine kinase inhibitors (TKI) by transcriptionally de-repressing platelet-derived growth factor receptor β (
a (GBM) cells develop resistance to anti-EGFR therapy. They demonstrate for the first time that an EGFR-dependent cancer can escape targeted therapy by developing dependence on another non-amplified, non-mutated RTK. Specifically, they show that GBMs with EGFR mutations evade EGFR tyrosine kinase inhibitors (TKI) by transcriptionally de-repressing platelet-derived growth factor receptor β (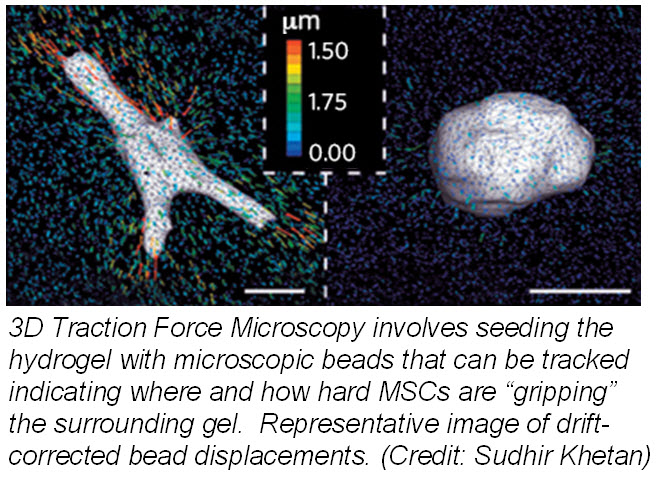
 ranslate that into chemical signals) can influence stem cell fate. Researchers from University of Pennsylvania showed that cell fate is regulated by cell-generated tension that is enabled through cell-mediated degradation of the covalently crosslinked matrix. When cultured on “softer” 2D covalently crosslinked gels (RGD-modified methacrylated hyaluronic acid hydrogels), mesenchymal stem cells (MSCs) differentiated into adipocytes when cultured in bipotential adipogenic/ osteogenic media. In contrast, MSCs cultured on “harder” 2D alginate gels differentiated into chondrocytes. This phenomenon was not present in 3D hydrogels and was attributed to the inability of cells to degrade the covalent cross-linked bonds resulting in MSCs differentiating into adipocytes. Introduction of proteolytically cleavable crosslinks and utilizing
ranslate that into chemical signals) can influence stem cell fate. Researchers from University of Pennsylvania showed that cell fate is regulated by cell-generated tension that is enabled through cell-mediated degradation of the covalently crosslinked matrix. When cultured on “softer” 2D covalently crosslinked gels (RGD-modified methacrylated hyaluronic acid hydrogels), mesenchymal stem cells (MSCs) differentiated into adipocytes when cultured in bipotential adipogenic/ osteogenic media. In contrast, MSCs cultured on “harder” 2D alginate gels differentiated into chondrocytes. This phenomenon was not present in 3D hydrogels and was attributed to the inability of cells to degrade the covalent cross-linked bonds resulting in MSCs differentiating into adipocytes. Introduction of proteolytically cleavable crosslinks and utilizing 
 Recently, a phase I clinical trial based on administration of autologous cardiosphere-derived cells (CDCs) to assess safety in patients with left ventricular dysfunction after MI was completed by Dr. Eduardo Marban atCedars-SinaiMedicalCenter[7]. The formation of cardiospheres was first described by Messina’s group who showed that human and mouse heart explants generated a layer of fibroblast-like cells over which small, phase-bright cells migrated, and once these phase-bright cells are transferred to non-adherent plates, they generate three-dimensional spherical structures [3]. Marban’s group modified Messina’s protocol by placing the cardiospheres in adherent plates where the cells begin to grow in monolayer, hence cardiosphere-derived cells, allowing for easier and faster expansion. Promising preclinical data which showed a reduction in infarct size and improved cardiac function after transplantation of CDCs in a porcine animal model prompted the phase I clinical trial [8].
Recently, a phase I clinical trial based on administration of autologous cardiosphere-derived cells (CDCs) to assess safety in patients with left ventricular dysfunction after MI was completed by Dr. Eduardo Marban atCedars-SinaiMedicalCenter[7]. The formation of cardiospheres was first described by Messina’s group who showed that human and mouse heart explants generated a layer of fibroblast-like cells over which small, phase-bright cells migrated, and once these phase-bright cells are transferred to non-adherent plates, they generate three-dimensional spherical structures [3]. Marban’s group modified Messina’s protocol by placing the cardiospheres in adherent plates where the cells begin to grow in monolayer, hence cardiosphere-derived cells, allowing for easier and faster expansion. Promising preclinical data which showed a reduction in infarct size and improved cardiac function after transplantation of CDCs in a porcine animal model prompted the phase I clinical trial [8].