
Francis Collins, the director of the National Institutes of Health’s Human Genome Research Institute, commented in a “Brave New Pharmacy” (Time Magazine, June 2001) that a new era of drug discovery was upon us where “if you understand the genetic basis of a disease, then you can predict what protein it produces and set about developing a drug to block it.” One such success is the development of Trastuzumab, an antibody against the extracellular domain of HER-2 (Human Epidermal Growth Factor Receptor 2 also known as ErbB-2) which was found to be over-expressed in 15-30% of breast cancers. However, targeting other ErbBs that are found in cancer has not been successful.
ErbB1 (also know as Epidermal Growth Factor Receptor or EGFR) has also been found to be over-expressed in a variety of tumors. EGFR is a 170,000 dalton transmembrane glycoprotein with intrinsic tyrosine kinase activity and family members include EGFR, ErbB2 (HER-2), ErbB3 and ErbB4. The predominant ligand for EGFR is epidermal growth factor (EGF), a 53-amino acid polypeptide, as well as the EGF family members transforming growth factor a (TGF-a), amphiregulin, heparin-binding EGF, β-cellulin, neuregulin and epiregulin. These proteins share a high binding affinity for EGFR and, upon binding to the receptor, induce EGFR dimerization, internalization and auto-phosphorylation which triggers signaling events involved in proliferation, migration, survival, and angiogenesis. Since EGFR signaling induces numerous mitogenic effects, EGFR over-expression and/ or gain-of-function mutations (EGFRvIII) can promote oncogenic transformation.
EGFR inhibitors have been developed to treat cancers that are caused by EGFR up-regulation such as breast, colorectal, head and neck, non-small cell lung carcinoma, pancreatic renal cell, squamous cell and thyroid cancer. EGFR inhibitors are either protein-tyrosine-kinase (PTK) inhibitors that bind to the tyrosine kinase domain or monoclonal antibodies that bind to the extracellular component of EGFR, preventing actual substrates from binding to the receptors and therefore preventing activation of EGFR. These drugs include Iressa (Gefitinib), Tarceva (Erlotinib), Erbitux (Cetuximab), Tykerb (Lapatinib), Vectibix (Panitumumab), and Caprelsa (Vandetanib).
However, there are numerous genetic mechanisms of resistance to anti-EGFR therapy including acquisition and/ or selection for secondary EGFR mutations, additional mutations in effectors resulting in constitutive activation of signaling pathways downstream of EGFR and co-occurrence of other amplified or mutated RTKs that bypass the EGFR pathway. EGFR mutations, which have been found in gliomas, non-small cell lung cancer, breast and ovarian cancer, have diminished response to EGFR therapy most likely due to conformational changes that affect intracellular domains involved in ATP binding sites. These mutations may also overwhelm the contribution of other signaling pathways for cell survival, thus allowing the cancer cells to increase their dependence on the EGFR signaling pathway for survival.
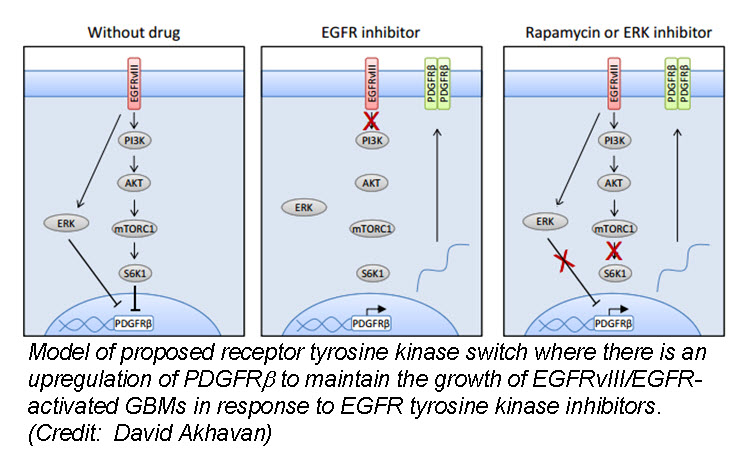
In the March issue of Cancer Discovery, a team of researchers identified a unique mechanism by which glioblastom a (GBM) cells develop resistance to anti-EGFR therapy. They demonstrate for the first time that an EGFR-dependent cancer can escape targeted therapy by developing dependence on another non-amplified, non-mutated RTK. Specifically, they show that GBMs with EGFR mutations evade EGFR tyrosine kinase inhibitors (TKI) by transcriptionally de-repressing platelet-derived growth factor receptor β (PDGFRβ). Cell lines, patient-derived tumor cultures, and xenotransplants showed that the persistently active EGFR mutation (EGFRvIII) suppressed PDGFRβ expression via mTORC1 and ERK-dependent mechanisms but that EGFR TKI treatment de-repressed PDGFRβ allowing the tumors to become “addicted” to a non-amplified, non-mutated RTK for continued growth and resistance to targeted treatment.
a (GBM) cells develop resistance to anti-EGFR therapy. They demonstrate for the first time that an EGFR-dependent cancer can escape targeted therapy by developing dependence on another non-amplified, non-mutated RTK. Specifically, they show that GBMs with EGFR mutations evade EGFR tyrosine kinase inhibitors (TKI) by transcriptionally de-repressing platelet-derived growth factor receptor β (PDGFRβ). Cell lines, patient-derived tumor cultures, and xenotransplants showed that the persistently active EGFR mutation (EGFRvIII) suppressed PDGFRβ expression via mTORC1 and ERK-dependent mechanisms but that EGFR TKI treatment de-repressed PDGFRβ allowing the tumors to become “addicted” to a non-amplified, non-mutated RTK for continued growth and resistance to targeted treatment.
Tumor tissue from GBM patients in a phase II clinical trial for an EGFR TKI (Lapatinib) revealed a reciprocal relationship between the activation of PDGFRβ and EGFRvIII. Tissue analysis from one patient before and after therapy revealed that Lapatinib treatment significantly reduced EGFR activation, but with a concomitant increase in PDGFRβ expression, supporting their in vitro and in vivo data that pharmacologic inhibition of EGFR results in RTK switching to PDGFRβ signaling.
We have targets and we have drugs, but RTK inhibitors have resulted in unfulfilled promises. Acquired drug resistance has presented a significant challenge for personalized cancer therapy. Despite being able to identify druggable RTK mutations in patients as well as second site mutations, non-genetic adaptive resistance mechanisms are able to “rewire” their circuitry through pathway crosstalk and release of inhibitory feedback loops. To further develop kinase cancer drugs, scientists need to combine RTK inhibitors with other agents (chemotherapy, radiation, other small molecules etc.) as well as target multiple tumor-promoting signaling pathways, either with drug combinations or with a single multi-targeted compound.
Further reading:
Akhavan D, Pourzia AL, Nourian AA, Williams KJ, Nathanson D, Babic I, Villa GR, Tanaka K, Nael A, Yang H, Dang J, Vinters HV, Yong WH, Flagg M, Tamanoi F, Sasayama T, James CD, Kornblum HI, Cloughesy TF, Cavenee WK, Bensinger SJ, Mischel PS. De-repression of PDGFRβ transcription promotes acquired resistance to EGFR tyrosine kinase inhibitors in glioblastoma patients. Cancer Discovery. 2013 Mar 27. [Epub ahead of print]
Deric L. Wheeler, Emily F. Dunn, and Paul M. Harari. Understanding resistance to EGFR inhibitors—impact on future treatment strategies. Nature Reviews Clinical Oncology. 2010 September; 7(9): 493–507.
James Perry, Masahiko Okamoto, Michael Guiou, Katsuyuki Shirai, Allison Errett, and Arnab Chakravarti. Novel Therapies in Glioblastoma. Neurology Research International. Volume 2012 (2012), Article ID 428565, 14 pages
 echanisms of acquired resistance to BRAF inhibitors (for detail please refer to my blog post titled ” RESISTANCE TO B-RAF INHIBITORS IN MELANOMA’’). Due to heterogeneous nature of cancer cells, it is crucial to gain a thorough understanding of the underlying drug-resistance mechanisms so that we can develop novel strategies to circumvent resistance and achieve more-prolonged responses. A recent study published in the journal Nature by Das Thakur and colleagues reported that intermittent treatment of vemurafenib prevented resistance in primary human melanoma xenografts. To study mechanisms of resistance to vemurafenib, Das Thakur et al. developed an animal model by continuously treating mice bearing a vemurafenib-naive, patient-derived BRAF-mutant melanoma with vemurafenib until drug resistance developed. Exome sequence analysis did not detect any secondary mutations in the coding sequences of BRAF, NRAS, KRAS, HRAS, and MEK1 in the resistant tumors. No alternatively spliced isoform of V600EBRAF, another known mechanism of vemurafenib resistance in melanoma was also detected in the resistant tumors. However, increased expression of V600EBRAF protein was noted in the resistant tumors and inhibition of V600EBRAF gene by RNA interference resulted in suppression of proliferation. These data suggested that the tumor cells were BRAF oncogene dependent and the observed drug resistance was due to the increased expression of V600EBRAF protein. In addition to these, another interesting observation was noted in this study when Das Thakur et al. tried to establish cell lines derived from the drug-resistant tumors. Cell lines derived from the drug-resistant tumors could not be developed without vemurafenib, where withdrawal of vemurafenib from the newly established cell lines changed cell morphology and decreased proliferation. This suggested that vemurafenib-resistant tumor cells in melanoma suffer a fitness deficit in the absence of vemurafenib. A similar type of vemurafenib dependency was also observed in SK-MEL239-C3 melanoma cells in which resistance is due to expression of a splice variant of V600EBRAF, and also in tumor cells isolated from a BRAF-mutated vemurafenib-resistant melanoma patient. Consistent with these findings, Das Thakur et al. observed tumor regression within 10 days in mice bearing vemurafenib resistant melanoma following cessation of vemurafenib treatment, although tumors eventually started re-growing. Collectively these results suggested that withdrawal of vemurafenib might create a hostile environment for drug-resistant cells and detain the onset of drug resistance. A comparison study made between continuous and intermittent vemurafenib treatment in human melanoma xenografts bearing mice further validated these observations. Drug resistance was developed in mice with 100 days receiving continuous treatment, whereas none of the mice on the intermittent treatment schedule exhibited drug resistance after 200 days of treatment. Therefore, these findings recommend that discontinuous treatment of vemurafenib may select against drug-resistant cells and prolong the responses to vemurafenib in melanoma. Future studies are needed especially in clinical trials to validate this proposal.
echanisms of acquired resistance to BRAF inhibitors (for detail please refer to my blog post titled ” RESISTANCE TO B-RAF INHIBITORS IN MELANOMA’’). Due to heterogeneous nature of cancer cells, it is crucial to gain a thorough understanding of the underlying drug-resistance mechanisms so that we can develop novel strategies to circumvent resistance and achieve more-prolonged responses. A recent study published in the journal Nature by Das Thakur and colleagues reported that intermittent treatment of vemurafenib prevented resistance in primary human melanoma xenografts. To study mechanisms of resistance to vemurafenib, Das Thakur et al. developed an animal model by continuously treating mice bearing a vemurafenib-naive, patient-derived BRAF-mutant melanoma with vemurafenib until drug resistance developed. Exome sequence analysis did not detect any secondary mutations in the coding sequences of BRAF, NRAS, KRAS, HRAS, and MEK1 in the resistant tumors. No alternatively spliced isoform of V600EBRAF, another known mechanism of vemurafenib resistance in melanoma was also detected in the resistant tumors. However, increased expression of V600EBRAF protein was noted in the resistant tumors and inhibition of V600EBRAF gene by RNA interference resulted in suppression of proliferation. These data suggested that the tumor cells were BRAF oncogene dependent and the observed drug resistance was due to the increased expression of V600EBRAF protein. In addition to these, another interesting observation was noted in this study when Das Thakur et al. tried to establish cell lines derived from the drug-resistant tumors. Cell lines derived from the drug-resistant tumors could not be developed without vemurafenib, where withdrawal of vemurafenib from the newly established cell lines changed cell morphology and decreased proliferation. This suggested that vemurafenib-resistant tumor cells in melanoma suffer a fitness deficit in the absence of vemurafenib. A similar type of vemurafenib dependency was also observed in SK-MEL239-C3 melanoma cells in which resistance is due to expression of a splice variant of V600EBRAF, and also in tumor cells isolated from a BRAF-mutated vemurafenib-resistant melanoma patient. Consistent with these findings, Das Thakur et al. observed tumor regression within 10 days in mice bearing vemurafenib resistant melanoma following cessation of vemurafenib treatment, although tumors eventually started re-growing. Collectively these results suggested that withdrawal of vemurafenib might create a hostile environment for drug-resistant cells and detain the onset of drug resistance. A comparison study made between continuous and intermittent vemurafenib treatment in human melanoma xenografts bearing mice further validated these observations. Drug resistance was developed in mice with 100 days receiving continuous treatment, whereas none of the mice on the intermittent treatment schedule exhibited drug resistance after 200 days of treatment. Therefore, these findings recommend that discontinuous treatment of vemurafenib may select against drug-resistant cells and prolong the responses to vemurafenib in melanoma. Future studies are needed especially in clinical trials to validate this proposal.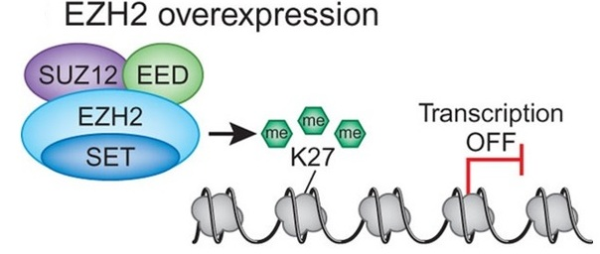
 d breast cancer. In both cases increased expression was found to be associated with tumor invasiveness, metastasis, and poor clinical outcome. In addition, elevated expression of EZH2 is also reported in several other tumors including gastric, lung, bladder, and endometrial cancer. Gain of functions as a result of acquired mutations in EZH2 was reported in lymphoma and meyloid neoplasms. The best characterized mechanism by which EZH2 exerts its oncogenic function is by transcriptional repression of genes via its histone methyltransferase activity. Genes which get transcriptionally repressed by EZH2 include tumor suppressor genes ARF, p57KIP2, FBXO32, p27, and BRCA1. In addition, this enzyme also activates transcription of gene CCND1 (cyclin D1) driving cell-cycle progression.
d breast cancer. In both cases increased expression was found to be associated with tumor invasiveness, metastasis, and poor clinical outcome. In addition, elevated expression of EZH2 is also reported in several other tumors including gastric, lung, bladder, and endometrial cancer. Gain of functions as a result of acquired mutations in EZH2 was reported in lymphoma and meyloid neoplasms. The best characterized mechanism by which EZH2 exerts its oncogenic function is by transcriptional repression of genes via its histone methyltransferase activity. Genes which get transcriptionally repressed by EZH2 include tumor suppressor genes ARF, p57KIP2, FBXO32, p27, and BRCA1. In addition, this enzyme also activates transcription of gene CCND1 (cyclin D1) driving cell-cycle progression.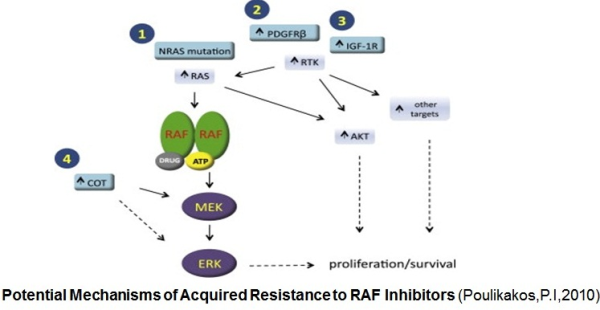
 ibitors. Activation of the MAPK signaling and increased expression of C-RAF (an isoform of B-RAF) was noted in melanoma cells resistant to B-RAF inhibitors. In this study Villanueva et al. (2010 ) also observed constitutive activation of the insulin-like growth factor receptor 1 (IFGR1), a receptor tyrosine kinase (RTK) in the resistant cells. As IGFR1 activates PI3K/Akt signaling, combined treatment of PI3K and MEK inhibitors resulted in resistance reversal. Increased levels of IGFR1 was also observed in melanoma patients failing vemurafenib suggesting activation of PI3K/Akt signaling via IGFR1 could limit the efficacy of B-RAF inhibitors in the clinic. Up-regulation of other RTKs was also found to be associated with the acquired resistance to vemurafenib. Tumor biopsies of melanoma patients failing vemurafenib exhibited over-expression of the platelet derived growth factor receptor-β (Nazarian et al., 2010).
ibitors. Activation of the MAPK signaling and increased expression of C-RAF (an isoform of B-RAF) was noted in melanoma cells resistant to B-RAF inhibitors. In this study Villanueva et al. (2010 ) also observed constitutive activation of the insulin-like growth factor receptor 1 (IFGR1), a receptor tyrosine kinase (RTK) in the resistant cells. As IGFR1 activates PI3K/Akt signaling, combined treatment of PI3K and MEK inhibitors resulted in resistance reversal. Increased levels of IGFR1 was also observed in melanoma patients failing vemurafenib suggesting activation of PI3K/Akt signaling via IGFR1 could limit the efficacy of B-RAF inhibitors in the clinic. Up-regulation of other RTKs was also found to be associated with the acquired resistance to vemurafenib. Tumor biopsies of melanoma patients failing vemurafenib exhibited over-expression of the platelet derived growth factor receptor-β (Nazarian et al., 2010).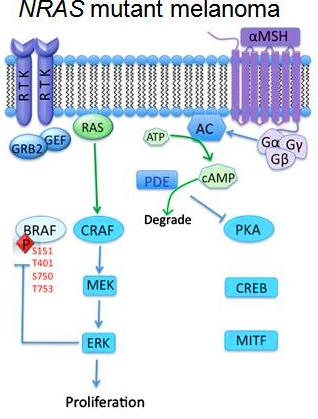


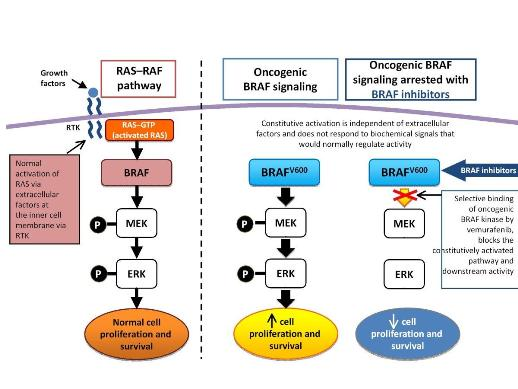
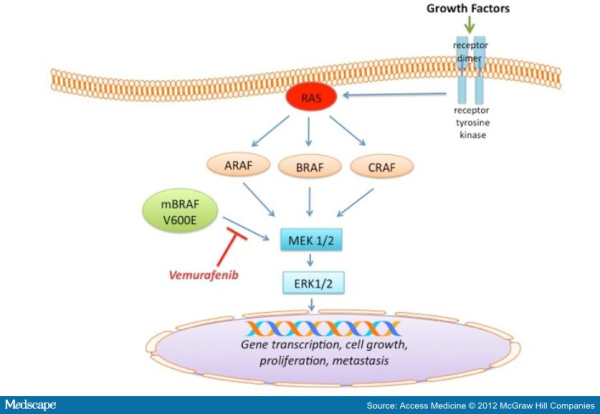
 growing Fibrosarcoma) protein is a serine/thereonine kinase. Three members of this kinase family are A-RAF, B-RAF, and C-RAF. These serine/threonine protein kinases, downstream of the membrane-bound small G protein RAS, are components of the mitogen activtated protein kinase (MAPK) signal transduction pathway. With closely overlapping functions, all members of the RAF family are associated with the activation of the MAPK pathway. Activation of the MAPK pathway has been associated with uncontrolled growth and drug resistance in several tumors. Researchers have identified over 50 distinct mutations in the B-RAF gene so far. However, most of these mutations are extremely rare. The most common mutation in melanoma, accounting for 90% of all B-RAF mutations, is the V600E mutation that occurs as a result of substitution of amino acid valine (V) to glutamic acid (E) at codon 600. Approximately 50% of melanomas harbor the V600E B-RAF mutation, while other mutations observed in melanomas are usually associated with the activation of N-RAS and c-KIT.
growing Fibrosarcoma) protein is a serine/thereonine kinase. Three members of this kinase family are A-RAF, B-RAF, and C-RAF. These serine/threonine protein kinases, downstream of the membrane-bound small G protein RAS, are components of the mitogen activtated protein kinase (MAPK) signal transduction pathway. With closely overlapping functions, all members of the RAF family are associated with the activation of the MAPK pathway. Activation of the MAPK pathway has been associated with uncontrolled growth and drug resistance in several tumors. Researchers have identified over 50 distinct mutations in the B-RAF gene so far. However, most of these mutations are extremely rare. The most common mutation in melanoma, accounting for 90% of all B-RAF mutations, is the V600E mutation that occurs as a result of substitution of amino acid valine (V) to glutamic acid (E) at codon 600. Approximately 50% of melanomas harbor the V600E B-RAF mutation, while other mutations observed in melanomas are usually associated with the activation of N-RAS and c-KIT.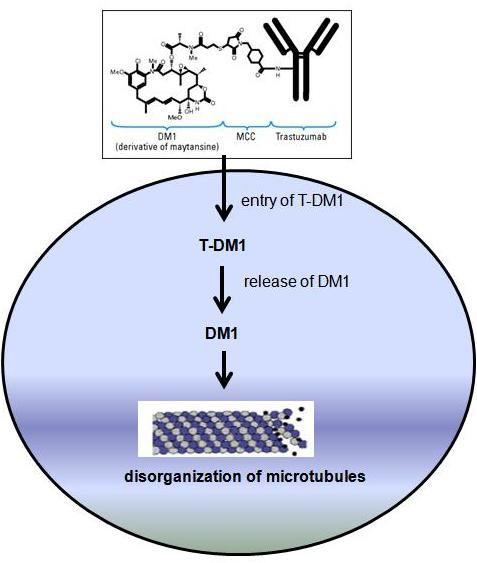
 HER-2 is a 185-kDa orphan transmembrane receptor tyrosine kinase. Dimerization of HER-2 with ligand- bound HER-3 or HER-4 receptor activates signaling pathways inside the cell. Activated HER-2 signaling stimulates cell proliferation and survival via activation of the MAPK and PI3K/Akt/mTOR pathways. Collectively these signaling pathways result in uncontrolled growth of the tumor. Several studies suggested that the overexpression/amplification of HER-2 may lead to the development and progression of pre-malignant breast disease and also tumor metastasis. Therefore, the association of HER-2 in breast cancer as well as its involvement in tumor aggressiveness makes this receptor an appropriate target for tumor-specific therapies. Several strategies have been developed to inhibit HER-2 signaling. These include a tyrosine kinase inhibitor called lapatinib and a recombinant humanized monoclonal antibody called trastuzumab (Herceptin®). In this post I will focus only on trastuzumab mediated therapy in breast cancer. Trastuzumab binds to the extracellular domain of the HER-2 receptor. This inhibits HER-2 signaling via MAPK and PI3K/Akt cascades. In addition, trastuzumab binding also increases membrane localization of the tumor suppressor gene phosphatase and tensin homolog (PTEN), and inhibitor of the PI3K/Aktpathway.
HER-2 is a 185-kDa orphan transmembrane receptor tyrosine kinase. Dimerization of HER-2 with ligand- bound HER-3 or HER-4 receptor activates signaling pathways inside the cell. Activated HER-2 signaling stimulates cell proliferation and survival via activation of the MAPK and PI3K/Akt/mTOR pathways. Collectively these signaling pathways result in uncontrolled growth of the tumor. Several studies suggested that the overexpression/amplification of HER-2 may lead to the development and progression of pre-malignant breast disease and also tumor metastasis. Therefore, the association of HER-2 in breast cancer as well as its involvement in tumor aggressiveness makes this receptor an appropriate target for tumor-specific therapies. Several strategies have been developed to inhibit HER-2 signaling. These include a tyrosine kinase inhibitor called lapatinib and a recombinant humanized monoclonal antibody called trastuzumab (Herceptin®). In this post I will focus only on trastuzumab mediated therapy in breast cancer. Trastuzumab binds to the extracellular domain of the HER-2 receptor. This inhibits HER-2 signaling via MAPK and PI3K/Akt cascades. In addition, trastuzumab binding also increases membrane localization of the tumor suppressor gene phosphatase and tensin homolog (PTEN), and inhibitor of the PI3K/Aktpathway. trastuzumab with taxanes docetaxel (Taxotere®) and paclitaxel (Taxol®) exhibited promising response in HER-2–overexpressing metastatic breast cancer.
trastuzumab with taxanes docetaxel (Taxotere®) and paclitaxel (Taxol®) exhibited promising response in HER-2–overexpressing metastatic breast cancer.
 RAS and BRAF oncogenes. In follicular thyroid carcinoma mutations of RAS oncogene in addition to other gene mutations was also observed. Aberrant activities of these oncogenes results in constitutive activation of the MAPK-pathway leading to inhibition of sodium-iodide symporter and thyroid peroxidase genes which participate in iodide uptake and thyroid hormone production respectively. In a pre-clinical study, Chakravarty et al. (2011) showed that mice with poorly differentiating thyroid cancer and overexpressing BRAF (V600E) oncogene failed to uptake RAI. Shutting BRAF activation off or inhibiting the MAPK-pathway with kinase inhibitors targeting BRAF or MEK rendered mice susceptible to a therapeutic dose of RAI. This suggests that inhibition of the MAPK-pathway and its associated protein kinases with the protein kinase inhibitors may facilitate RAI uptake in refractory thyroid cancer patients harboring MAPK-pathway activation.
RAS and BRAF oncogenes. In follicular thyroid carcinoma mutations of RAS oncogene in addition to other gene mutations was also observed. Aberrant activities of these oncogenes results in constitutive activation of the MAPK-pathway leading to inhibition of sodium-iodide symporter and thyroid peroxidase genes which participate in iodide uptake and thyroid hormone production respectively. In a pre-clinical study, Chakravarty et al. (2011) showed that mice with poorly differentiating thyroid cancer and overexpressing BRAF (V600E) oncogene failed to uptake RAI. Shutting BRAF activation off or inhibiting the MAPK-pathway with kinase inhibitors targeting BRAF or MEK rendered mice susceptible to a therapeutic dose of RAI. This suggests that inhibition of the MAPK-pathway and its associated protein kinases with the protein kinase inhibitors may facilitate RAI uptake in refractory thyroid cancer patients harboring MAPK-pathway activation. resistant to RAI. Selumetinib is currently in clinical trials for various solid and hematologic malignancies. Among 24 patients screened for the study 5 had NRAS-mutant tumors. All of them showed augmented RAI uptake following treatment with selumetinib; 4 patients exhibited confirmed partial responses (PR) and in 1 patient no disease progression was noted following RAI treatment. No significant levels of toxic effects to selumetinib were observed in this study. Increased RAI uptake and confirmed PR was also observed in 1 patient with BRAF mutation after treatment with selumetinib. However, selumetinib treatment in majority of the patients with BRAF mutations enrolled in this study did not increase RAI uptake up to the threshold level required for therapy. Therefore, further studies are required to understand the differences observed between RAS-mutant and BRAF-mutant tumors.
resistant to RAI. Selumetinib is currently in clinical trials for various solid and hematologic malignancies. Among 24 patients screened for the study 5 had NRAS-mutant tumors. All of them showed augmented RAI uptake following treatment with selumetinib; 4 patients exhibited confirmed partial responses (PR) and in 1 patient no disease progression was noted following RAI treatment. No significant levels of toxic effects to selumetinib were observed in this study. Increased RAI uptake and confirmed PR was also observed in 1 patient with BRAF mutation after treatment with selumetinib. However, selumetinib treatment in majority of the patients with BRAF mutations enrolled in this study did not increase RAI uptake up to the threshold level required for therapy. Therefore, further studies are required to understand the differences observed between RAS-mutant and BRAF-mutant tumors.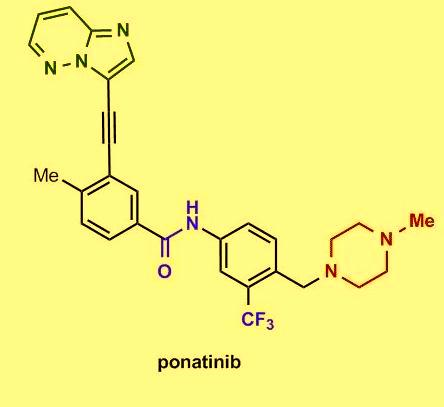
 Several in vitro studies showed that the tyrosine kinase chimeric protein Bcr-Abl encoded by the BCR-ABL gene is constitutively active in leukemia cells and has oncogenic properties. Bcr-Abl chimeric protein has been found to be associated with genomic instability and thereby suggested to be responsible for progression to advanced phases of CML.
Several in vitro studies showed that the tyrosine kinase chimeric protein Bcr-Abl encoded by the BCR-ABL gene is constitutively active in leukemia cells and has oncogenic properties. Bcr-Abl chimeric protein has been found to be associated with genomic instability and thereby suggested to be responsible for progression to advanced phases of CML. Among chronic-phase CML patients with T315I mutation 100% exhibited hematologic response, 92% had a major cytogenetic response, 75% exhibited complete cytogenetic response, and 67% had a major molecular response. The most common side effects reported in the study include hypertension, rash, abdominal pain, fatigue, headache, dry skin, constipation, fever, joint pain, and nausea. Clinically promising similar results were also observed in the PACE trial, a multicenter, international, single-arm clinical trial of 449 patients with disease that was resistant or intolerant to prior tyrosine kinase inhibitor therapy. On December 14, 2012, the FDA approved ponatinib (Iclusig tablets) for the treatment of adult patients with all phases of CML that are resistant or intolerant to prior tyrosine kinase inhibitor therapy.
Among chronic-phase CML patients with T315I mutation 100% exhibited hematologic response, 92% had a major cytogenetic response, 75% exhibited complete cytogenetic response, and 67% had a major molecular response. The most common side effects reported in the study include hypertension, rash, abdominal pain, fatigue, headache, dry skin, constipation, fever, joint pain, and nausea. Clinically promising similar results were also observed in the PACE trial, a multicenter, international, single-arm clinical trial of 449 patients with disease that was resistant or intolerant to prior tyrosine kinase inhibitor therapy. On December 14, 2012, the FDA approved ponatinib (Iclusig tablets) for the treatment of adult patients with all phases of CML that are resistant or intolerant to prior tyrosine kinase inhibitor therapy.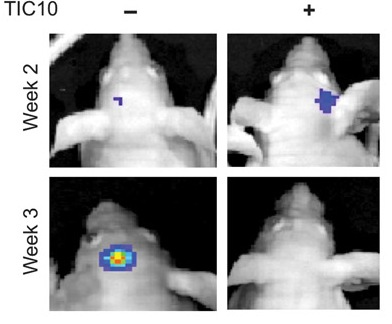
 R4) and TR2 (DR5). TRAIL also binds with two other receptors TR3 and TR4. Unlike TR1 and TR2, these receptors have incomplete death domains. Elevated expression of TR3 and TR4 in normal cells is thereby suggested to protect normal cells from TRAIL-induced death signaling. Induction of apoptotic signaling involves binding of TRAIL to DR4 or DR5. This results in homotrimerization and activation of receptors, enabling the receptors’ death domain to recruit the adaptor protein Fas-associated death domain along with pro-caspase- 8 and pro-caspase- 10. These all together then form the multi-protein death-inducing signaling complex, (DISC). Inside the DISC procaspeses become autoactivated and become caspase-8/10. Activated caspase-8/10 then activates downstream effector caspase-3 or caspase-7. Finally, cleavage of downstream substrates by effector caspases results in DNA fragmentation leading to apoptosis.
R4) and TR2 (DR5). TRAIL also binds with two other receptors TR3 and TR4. Unlike TR1 and TR2, these receptors have incomplete death domains. Elevated expression of TR3 and TR4 in normal cells is thereby suggested to protect normal cells from TRAIL-induced death signaling. Induction of apoptotic signaling involves binding of TRAIL to DR4 or DR5. This results in homotrimerization and activation of receptors, enabling the receptors’ death domain to recruit the adaptor protein Fas-associated death domain along with pro-caspase- 8 and pro-caspase- 10. These all together then form the multi-protein death-inducing signaling complex, (DISC). Inside the DISC procaspeses become autoactivated and become caspase-8/10. Activated caspase-8/10 then activates downstream effector caspase-3 or caspase-7. Finally, cleavage of downstream substrates by effector caspases results in DNA fragmentation leading to apoptosis. gen-activated protein kinase (MAPK) pathways was noted following TIC10 treatment in this in vivo study that resulted in translocation of the transcription factor Foxo3a into the nucleus, where Foxo3a induced expression of TRAIL gene to activate apoptosis.
gen-activated protein kinase (MAPK) pathways was noted following TIC10 treatment in this in vivo study that resulted in translocation of the transcription factor Foxo3a into the nucleus, where Foxo3a induced expression of TRAIL gene to activate apoptosis.