
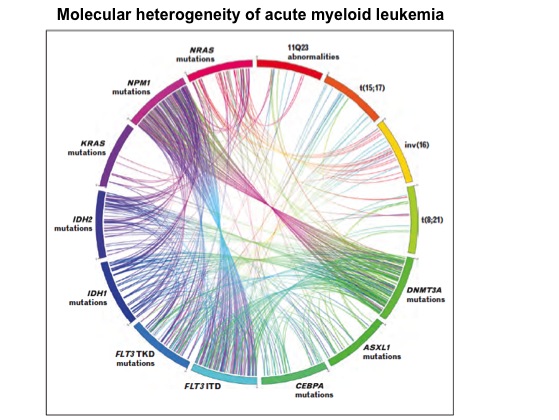
Acute myeloid leukemia (AML), a cancer of hematopoietic cells, is a molecularly heterogeneous disease. AML is associated with several genetic changes that alter normal hematopoietic growth and differentiation, resulting in the accumulation of large numbers of abnormal immature myeloid cells in the bone marrow and peripheral blood. These cells are capable of dividing and proliferating, but cannot differentiate into mature hematopoietic cells. Recurrent structural alterations of chromosomes are the established diagnostic and prognostic markers in AML. Several studies using targeted sequencing (determination of DNA sequence of specific areas of interest within the genome) identified recurrent gene mutations that contained diagnostic and prognostic information, including mutations in FLT3, NPM1, KIT, CEBPA, and TET2 genes. In addition, massively parallel sequencing (high-throughput approaches of DNA sequencing, also called next-generation sequencing) has discovered recurrent mutations in DNMT3A and IDH1/2 genes that may also provide prognostic information for some patients. Even though these genetic abnormalities may play an essential role in the pathogenesis of AML, nearly 50% of AML patients have normal karyotype (an organized profile of a person’s chromosomes). Based on the cytogenetic analysis, AML patients are classified into three major risk categories: favorable, intermediate and unfavorable. AML patients with PML-RARA, RUNX1-RUNX1T1, or MYTH11-CBFB gene fusions (as a result of chromosomal rearrangements) profile belong to favorable- risk category and have shown relatively good response to chemotherapy. Patients with complex genetic alterations (e.g. monosomy karyotype) are categorized into unfavorable-risk profile. Majority of the AML patients exhibit normal karyotype and belong to intermediate-risk category. Some of these patients respond well to chemotherapy while others don’t. Since nearly 50% of AML patients have normal chromosomal profile, better molecular characterization of pathogenesis of AML is required for better approaches to therapy. In addition, next-generation sequencing (NGS) studies have revealed that even though AML usually harbor hundreds of mutated genes, only a limited number of mutated genes serve as driver mutations (i.e. causing the tumor). Among the different adult cancer types sequenced extensively so far, AML has had the fewest mutations discovered. Therefore, identification of a significant number of novel driver mutations present at low frequency in AML will help to better understand the leukemogenesis.
Based on the cytogenetic analysis, AML patients are classified into three major risk categories: favorable, intermediate and unfavorable. AML patients with PML-RARA, RUNX1-RUNX1T1, or MYTH11-CBFB gene fusions (as a result of chromosomal rearrangements) profile belong to favorable- risk category and have shown relatively good response to chemotherapy. Patients with complex genetic alterations (e.g. monosomy karyotype) are categorized into unfavorable-risk profile. Majority of the AML patients exhibit normal karyotype and belong to intermediate-risk category. Some of these patients respond well to chemotherapy while others don’t. Since nearly 50% of AML patients have normal chromosomal profile, better molecular characterization of pathogenesis of AML is required for better approaches to therapy. In addition, next-generation sequencing (NGS) studies have revealed that even though AML usually harbor hundreds of mutated genes, only a limited number of mutated genes serve as driver mutations (i.e. causing the tumor). Among the different adult cancer types sequenced extensively so far, AML has had the fewest mutations discovered. Therefore, identification of a significant number of novel driver mutations present at low frequency in AML will help to better understand the leukemogenesis.
A study recently published in the New England Journal of Medicine (May 1st, 2013) by researchers at The Cancer Genome Atlas (TCGA) group led by Timothy J. Ley broadly classified the genomic alterations that frequently underlie the development of AML. This study also suggested potential new drug targets and treatment strategies for AML. In this study the genome of 200 newly diagnosed adult cases of AML patients, representing all of the known subtypes, was analyzed by performing whole-genome sequencing (50 cases) and whole-exome sequencing (150 cases). In addition, RNA and micro-RNA sequencing and DNA-methylation analysis was also performed.
Each AML genome was compared to the  normal genome derived from a skin sample of the same patient. The recurrently mutated genes discovered in this study were grouped into nine categories that were defined according to biologic function and that are considered to play a role in AML pathogenesis. Some of these groups include: tumor suppressor genes, transcription-factor fusions, activated signaling genes and epigenetic modifiers (DNA-methylation related genes and chromatin-modifying genes) with the latter being the most frequently mutated class of genes found in this study. At least one potential driver mutation was identified in nearly all AML samples including genes that are well established as being associated with AML pathogenesis (eg. FLT3, NPM1, DNMT3A, IDH1, IDH2, and CEBPA).
normal genome derived from a skin sample of the same patient. The recurrently mutated genes discovered in this study were grouped into nine categories that were defined according to biologic function and that are considered to play a role in AML pathogenesis. Some of these groups include: tumor suppressor genes, transcription-factor fusions, activated signaling genes and epigenetic modifiers (DNA-methylation related genes and chromatin-modifying genes) with the latter being the most frequently mutated class of genes found in this study. At least one potential driver mutation was identified in nearly all AML samples including genes that are well established as being associated with AML pathogenesis (eg. FLT3, NPM1, DNMT3A, IDH1, IDH2, and CEBPA).
This study was the first to observe recurrent mutations in cohesin genes, which are important in cell division, in 13% of cases of AML samples. In addition, this study also observed a mutation in microRNA 142 (miR-142). Overall this study provided a detailed understanding of the genetic and epigenetic changes associated with adult de novo AML. Future studies are warranted to understand the relationship between these alterations and treatment results.
References:
1. Bacher U, Schnittger S, Haferlach T. Molecular genetics in acute myeloid leukemia. Curr Opin Oncol. 2010;22:646-55.
2. Stirewalt DL, Radich JP. The role of FLT3 in haematopoietic malignancies. Nat Rev Cancer. 2003;3:650-65.
3. Yamashita Y, Yuan J, Suetake I, Suzuki H, Ishikawa Y, Choi YL, et al. Array-based genomic resequencing of human leukemia. Oncogene. 2010;29:3723-31.
4. Network TCGAR. Genomic and Epigenomic Landscapes of Adult De Novo Acute Myeloid Leukemia. N Engl J Med. 2013.