
Francis Collins, the director of the National Institutes of Health’s Human Genome Research Institute, commented in a “Brave New Pharmacy” (Time Magazine, June 2001) that a new era of drug discovery was upon us where “if you understand the genetic basis of a disease, then you can predict what protein it produces and set about developing a drug to block it.” One such success is the development of Trastuzumab, an antibody against the extracellular domain of HER-2 (Human Epidermal Growth Factor Receptor 2 also known as ErbB-2) which was found to be over-expressed in 15-30% of breast cancers. However, targeting other ErbBs that are found in cancer has not been successful.
ErbB1 (also know as Epidermal Growth Factor Receptor or EGFR) has also been found to be over-expressed in a variety of tumors. EGFR is a 170,000 dalton transmembrane glycoprotein with intrinsic tyrosine kinase activity and family members include EGFR, ErbB2 (HER-2), ErbB3 and ErbB4. The predominant ligand for EGFR is epidermal growth factor (EGF), a 53-amino acid polypeptide, as well as the EGF family members transforming growth factor a (TGF-a), amphiregulin, heparin-binding EGF, β-cellulin, neuregulin and epiregulin. These proteins share a high binding affinity for EGFR and, upon binding to the receptor, induce EGFR dimerization, internalization and auto-phosphorylation which triggers signaling events involved in proliferation, migration, survival, and angiogenesis. Since EGFR signaling induces numerous mitogenic effects, EGFR over-expression and/ or gain-of-function mutations (EGFRvIII) can promote oncogenic transformation.
EGFR inhibitors have been developed to treat cancers that are caused by EGFR up-regulation such as breast, colorectal, head and neck, non-small cell lung carcinoma, pancreatic renal cell, squamous cell and thyroid cancer. EGFR inhibitors are either protein-tyrosine-kinase (PTK) inhibitors that bind to the tyrosine kinase domain or monoclonal antibodies that bind to the extracellular component of EGFR, preventing actual substrates from binding to the receptors and therefore preventing activation of EGFR. These drugs include Iressa (Gefitinib), Tarceva (Erlotinib), Erbitux (Cetuximab), Tykerb (Lapatinib), Vectibix (Panitumumab), and Caprelsa (Vandetanib).
However, there are numerous genetic mechanisms of resistance to anti-EGFR therapy including acquisition and/ or selection for secondary EGFR mutations, additional mutations in effectors resulting in constitutive activation of signaling pathways downstream of EGFR and co-occurrence of other amplified or mutated RTKs that bypass the EGFR pathway. EGFR mutations, which have been found in gliomas, non-small cell lung cancer, breast and ovarian cancer, have diminished response to EGFR therapy most likely due to conformational changes that affect intracellular domains involved in ATP binding sites. These mutations may also overwhelm the contribution of other signaling pathways for cell survival, thus allowing the cancer cells to increase their dependence on the EGFR signaling pathway for survival.
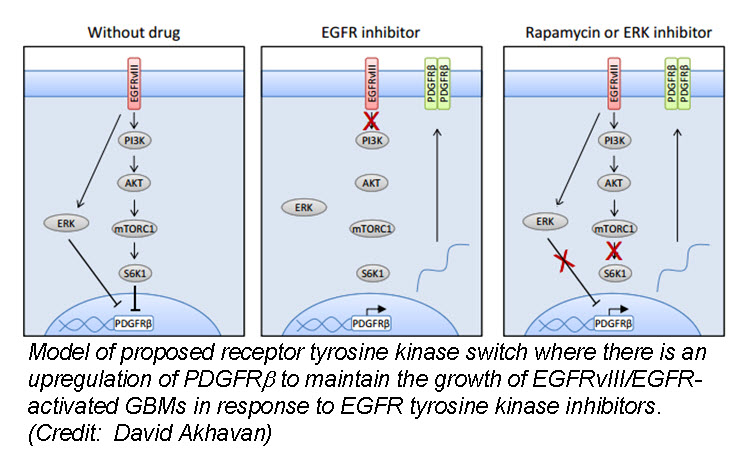
In the March issue of Cancer Discovery, a team of researchers identified a unique mechanism by which glioblastom a (GBM) cells develop resistance to anti-EGFR therapy. They demonstrate for the first time that an EGFR-dependent cancer can escape targeted therapy by developing dependence on another non-amplified, non-mutated RTK. Specifically, they show that GBMs with EGFR mutations evade EGFR tyrosine kinase inhibitors (TKI) by transcriptionally de-repressing platelet-derived growth factor receptor β (PDGFRβ). Cell lines, patient-derived tumor cultures, and xenotransplants showed that the persistently active EGFR mutation (EGFRvIII) suppressed PDGFRβ expression via mTORC1 and ERK-dependent mechanisms but that EGFR TKI treatment de-repressed PDGFRβ allowing the tumors to become “addicted” to a non-amplified, non-mutated RTK for continued growth and resistance to targeted treatment.
a (GBM) cells develop resistance to anti-EGFR therapy. They demonstrate for the first time that an EGFR-dependent cancer can escape targeted therapy by developing dependence on another non-amplified, non-mutated RTK. Specifically, they show that GBMs with EGFR mutations evade EGFR tyrosine kinase inhibitors (TKI) by transcriptionally de-repressing platelet-derived growth factor receptor β (PDGFRβ). Cell lines, patient-derived tumor cultures, and xenotransplants showed that the persistently active EGFR mutation (EGFRvIII) suppressed PDGFRβ expression via mTORC1 and ERK-dependent mechanisms but that EGFR TKI treatment de-repressed PDGFRβ allowing the tumors to become “addicted” to a non-amplified, non-mutated RTK for continued growth and resistance to targeted treatment.
Tumor tissue from GBM patients in a phase II clinical trial for an EGFR TKI (Lapatinib) revealed a reciprocal relationship between the activation of PDGFRβ and EGFRvIII. Tissue analysis from one patient before and after therapy revealed that Lapatinib treatment significantly reduced EGFR activation, but with a concomitant increase in PDGFRβ expression, supporting their in vitro and in vivo data that pharmacologic inhibition of EGFR results in RTK switching to PDGFRβ signaling.
We have targets and we have drugs, but RTK inhibitors have resulted in unfulfilled promises. Acquired drug resistance has presented a significant challenge for personalized cancer therapy. Despite being able to identify druggable RTK mutations in patients as well as second site mutations, non-genetic adaptive resistance mechanisms are able to “rewire” their circuitry through pathway crosstalk and release of inhibitory feedback loops. To further develop kinase cancer drugs, scientists need to combine RTK inhibitors with other agents (chemotherapy, radiation, other small molecules etc.) as well as target multiple tumor-promoting signaling pathways, either with drug combinations or with a single multi-targeted compound.
Further reading:
Akhavan D, Pourzia AL, Nourian AA, Williams KJ, Nathanson D, Babic I, Villa GR, Tanaka K, Nael A, Yang H, Dang J, Vinters HV, Yong WH, Flagg M, Tamanoi F, Sasayama T, James CD, Kornblum HI, Cloughesy TF, Cavenee WK, Bensinger SJ, Mischel PS. De-repression of PDGFRβ transcription promotes acquired resistance to EGFR tyrosine kinase inhibitors in glioblastoma patients. Cancer Discovery. 2013 Mar 27. [Epub ahead of print]
Deric L. Wheeler, Emily F. Dunn, and Paul M. Harari. Understanding resistance to EGFR inhibitors—impact on future treatment strategies. Nature Reviews Clinical Oncology. 2010 September; 7(9): 493–507.
James Perry, Masahiko Okamoto, Michael Guiou, Katsuyuki Shirai, Allison Errett, and Arnab Chakravarti. Novel Therapies in Glioblastoma. Neurology Research International. Volume 2012 (2012), Article ID 428565, 14 pages