
The blood-brain barrier (BBB) is a highly complex structure of the cerebral vasculature; it functions as a selective filter for certain molecules entering and exiting the brain. Through regulating the exchange of very specific molecules and cells between the central nervous system’s Cerebral Spinal Fluid (CSF) and the peripheral blood of the circulatory system, the BBB protects the brain and maintains the scrupulous environment required for neurons and other glial cells to function properly. The significant consequence of BBB disruption is the increased permeability, leading to extravasation of circulating peripheral blood mononuclear cells (PBMC), along with other unspecific molecules. Thus, BBB dysfunction plays a major role in a wide range of neurologic conditions, including Multiple Sclerosis (MS) and Alzheimer’s disease (AD).
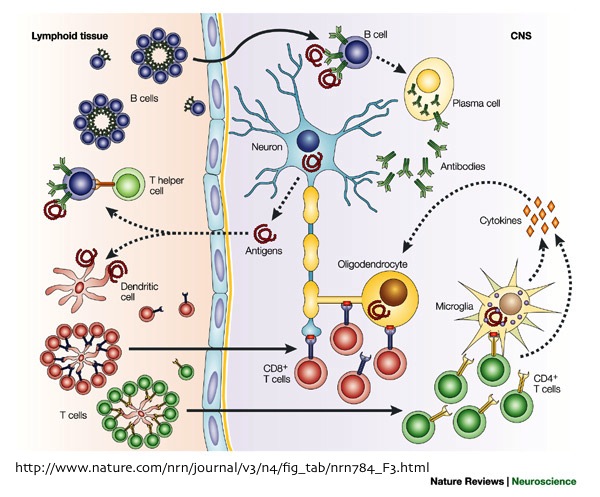
MS is associated with the infiltration of CD4+ and CD8+ T-cells and B-cells within the acute inflammatory lesions or the areas of demyelination. The presence of these immune cells at these locations, indicate alterations in BBB structure, which allowed their crossing into the central nervous system (CNS).
During MS, perivascular lesions are formed by T-cells entering the CNS and releasing cytokines that cause inflammation of the endothelial lining (cells forming the blood vessels). This inflammatory response stimulates vasculature in MS lesions to express several cell adhesion molecules (CAMs). The interaction between these CAMs and their leukocytic integrins, permit immune cells to adhere to the inflamed cerebral vasculature, cross the BBB, and proceed to infiltrate the parenchyma. This inflammatory reaction is a vicious cycle; as more immune cells are activated within the CNS, the inflammation amplifies, leading to further BBB damage and recruitment of additional lymphocytes, such as B-cells and cytotoxic T-cells, responsible for the eventual activation of associated macrophages to destroy Myelin. In addition, it has been reported that the firm attachment of monocytes to endothelial cells, stimulates monocytes to secrete reactive oxygen species (ROS) that may further increase BBB permeability to T-cells and macrophages.
The initial stimulant of MS is not yet clear. Nonetheless, many recent studies suggest that BBB disruption is an early event in MS lesion formation, followed by the massive infiltration of immune cells, which proceed to destroy the myelin and damage the oligodendrocytes.

Several cerebrovascular abnormalities, such as damaged endothelial and pericyte, microvascular degeneration, reduced glucose transport across the BBB, and abnormal expression of inflammatory markers in the cerebral vasculature have been described in Alzheimer’s disease patients. These observations are associated to the deposition of the β-amyloid peptide (Aβ) in the walls of the BBB vasculature which lead to the loss of certain tight junction proteins that result in disruption of the BBB and eventual cerebral neuroinflammation through activating microglia.
The two pathological hallmarks of AD are: 1) the increased production and accumulation of amyloid-β peptides (Aβ)- derived from amyloid precursor protein (APP)- forming neuritic/senile plaques in the brain tissue, 2) the intraneuronal neurofibrillary tangles (NFTs) composed of hyperphosphorylated tau protein, which lead to the loss of synapses and neurons in affected regions. Consequently, these factors in addition to a reduction of Aβ clearance from the brain, which leads to its extracellular accumulation, and its direct toxic effects, induce the subsequent activation of microglial cells; this inflammatory response results in the release of numerous inflammatory neurotoxic cytokines that will disrupt the neurons’ cytoskeleton, leading to their dysfunction and eventual cellular death.

Recent studies suggest that transforming growth factor-β1 (TGF-β1) may play a role in BBB dysfunction in AD pathogenesis. TGF-β1 has a constitutive role in the suppression of inflammation and control microglial activation in the CNS. These observations indicate a significant impairment of TGF-β1 signaling in AD brain. Furthermore, astrocytes may be reactivated by TGF-β1, which would result in further production of Aβ and undergo the aggravating astrogliosis.
Although it is not yet clear whether these vascular changes are an initial cause for development of AD or if they take place during the later stages of the disease, most Alzheimer’s patients exhibit vascular pathology and develop intracerebral hemorrhage and cerebral infracts.
In conclusion, there is strong evidence pointing to the involvement of the BBB dysfunction in many neurologic disorders including MS and AD. Moreover, one common denominator of the BBB malfunction in these diseases is the close association of the astrocytes activated by the interactions between the immune cells and the cerebral vasculature, leading to further increased permeability of BBB. However, further investigations must focus on identifying the mechanisms responsible for the initial cause of the the inflammatory cascade, and the destructive interactions between reactivated astorcytes and endothelial cells lining the BBB.
Further Reading:
Current and future treatments for Alzheimer’s disease. Yiannopoulou KG, PapageorgiouSG. (2013) Ther Adv Neurol Disord, 6(1) 19–33.
Inflammatory events at blood–brain barrier in neuroinflammatory and neurodegenerative disorders: Implications for clinical disease. de Vries HE, Kooij G, Frenkel D, Georgopoulos S, Monsonego A, Janigro D. (2012) Epilepsia, 53(Suppl. 6):45–52.
New concepts in the immunopathogenesis of multiple sclerosis. . (April 2002) Nature Reviews Neuroscience, 3, 291-301.
Neuroinflammation and blood–brain barrier changes in capillary amyloid angiopathy.Carrano A, Hoozemans JJ, van der Vies SM, van Horssen J, de Vries HE, Rozemuller AJ. (2012) Neurodegener Dis, 10:329– 331.
