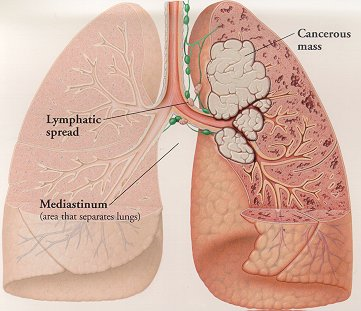
Prostate cancer (PCa) is the second most common cause of male cancer-related death. According to the National Cancer Institute, in the United States in 2013 the estimated new cases of prostate cancer would be 238,590 and deaths would be 29,720.
Bone metastases are a serious problem in men with advanced PCa. Bone metastases increase the risk of skeletal-related events (SREs) which include pathological fractures, spinal cord compression, bone pain. Both bone metastases and SREs are associated with an unfavorable prognosis and greatly affect quality of life.
Numerous studies have shown the importance of androgens (steroid hormones) in the development of PCa (although the exact role of androgen in PCa development is yet to be determined). Therefore, continuous androgen deprivation has been the standard therapy for metastatic hormone-sensitive disease. Despite a high response rate, resistance to androgen-deprivation therapy occurs in most patients, resulting in a median survival of 2.5 to 3 years. Thus, resulting in the development of castration resistant (CRPC) or hormone-refractory (HRPC) stage. Standard chemotherapy has not proven to be very effective in cases of metastatic CRPC, with a 10–20% response rate and approximately one-year median survival. In the United States docetaxel and cabazitaxel are the only Food and Drug Administration (FDA)-approved chemotherapies for the treatment of metastatic CRPC. Even though these drugs palliate symptoms, the overall survival benefit is moderate. In addition, a cellular immunotherapeutic agent sipuleucel-T (Provenge; Dendreon Corp) has been shown to increase overall survival period by 4.1 months on average but not progression-free survival time for patients with metastatic CRPC.
On May 15th, 2013, the U.S. FDA approved  radium Ra 223 dichloride (Xofigo®; Bayer HealthCare Pharmaceuticals) to treat men with metastatic castration-resistant prostate cancer with bone metastases after receiving medical or surgical treatment. The efficacy of Xofigo® was evaluated in a single clinical trial (Phase 3 ALSYMPCA trial) of 809 men with metastatic castration-resistant prostate cancer. Compared to the controls (patients received placebo plus standard care) with median survival of 11.2 months, patients who received Xofigo® lived a median of 14 months. The side effects noted during the clinical trials among patients treated with Xofigo® were nausea, diarrhea, vomiting, and swelling of the leg, foot, or ankle.
radium Ra 223 dichloride (Xofigo®; Bayer HealthCare Pharmaceuticals) to treat men with metastatic castration-resistant prostate cancer with bone metastases after receiving medical or surgical treatment. The efficacy of Xofigo® was evaluated in a single clinical trial (Phase 3 ALSYMPCA trial) of 809 men with metastatic castration-resistant prostate cancer. Compared to the controls (patients received placebo plus standard care) with median survival of 11.2 months, patients who received Xofigo® lived a median of 14 months. The side effects noted during the clinical trials among patients treated with Xofigo® were nausea, diarrhea, vomiting, and swelling of the leg, foot, or ankle.
The alpha particle-emitting pharmaceutical Xofigo® is a radio-therapeutic drug. It mimics calcium and forms complexes with the bone mineral hydroxyapatite at areas of increased bone turnover, such as bone metastases. This drug is administered as an intravenous injection.
Overall, Xofigo® was found to extend the survival of men with metastatic prostate cancer and expected to be available in the clinic within a few weeks.
References:
1. Yagoda A, Petrylak D: Cytotoxic chemotherapy for advanced hormone-resistant prostate cancer. Cancer 1993, 71(3 Suppl):1098-1109.
2. Harrison MR, Wong TZ, Armstrong AJ, George DJ: Radium-223 chloride: a potential new treatment for castration-resistant prostate cancer patients with metastatic bone disease. Cancer Manag Res 2013, 5:1-14.
3. Karantanos T, Corn PG, Thompson TC: Prostate cancer progression after androgen deprivation therapy: mechanisms of castrate resistance and novel therapeutic approaches. Oncogene 2013.










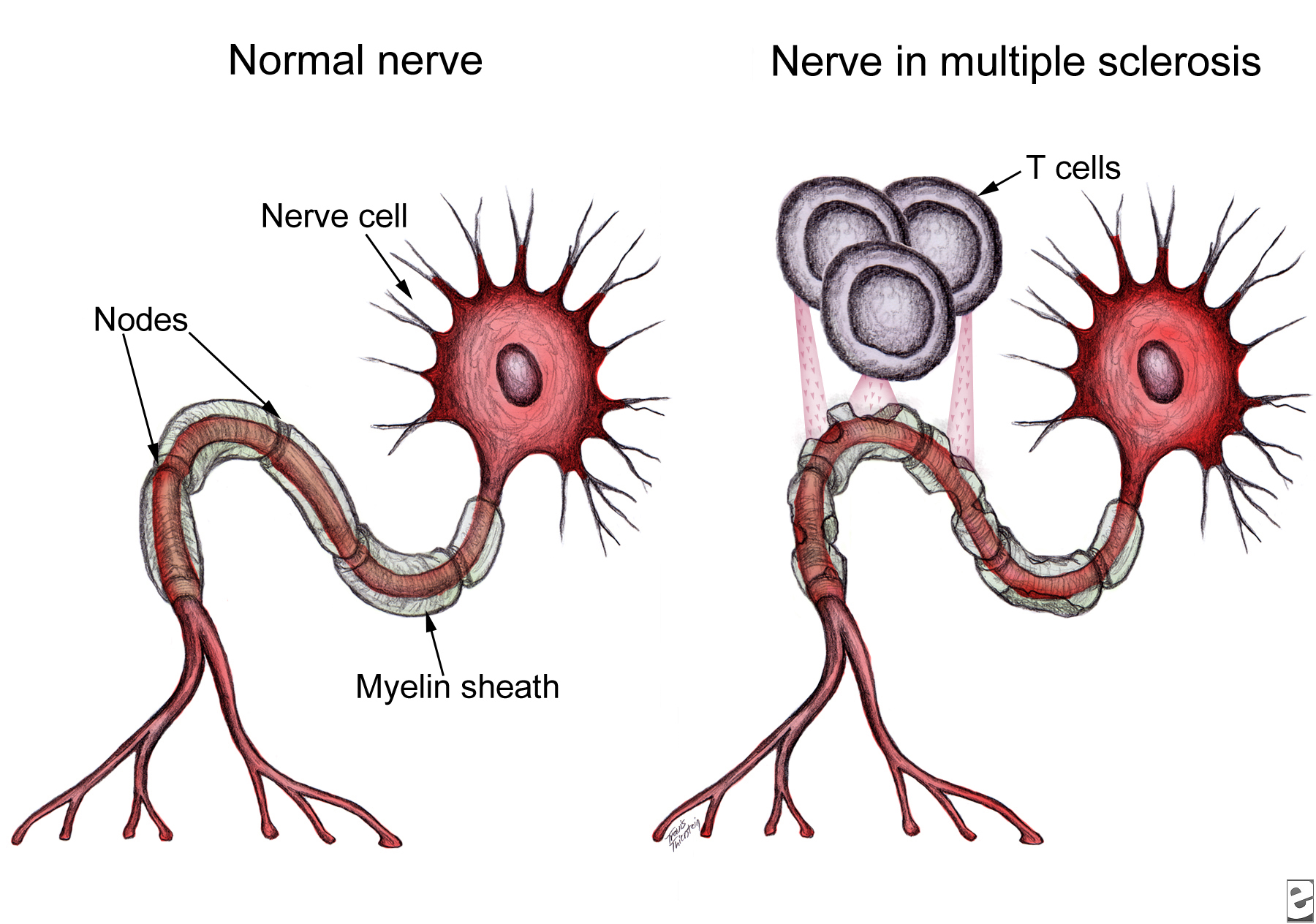

 These results support the epitope-spreading hypothesis, which indicates that MS patients make antibodies against one or a few myelin proteins, but as the disease progresses, the autoimmune response spreads to other myelin sheath epitopes. In their recent publication, Lutterotti et al. provide sufficient evidence necessitating emphasis not only on the specific target antigens, but also on the facility to inhibit epitope spreading, preferably prior to diversification of the
These results support the epitope-spreading hypothesis, which indicates that MS patients make antibodies against one or a few myelin proteins, but as the disease progresses, the autoimmune response spreads to other myelin sheath epitopes. In their recent publication, Lutterotti et al. provide sufficient evidence necessitating emphasis not only on the specific target antigens, but also on the facility to inhibit epitope spreading, preferably prior to diversification of the