
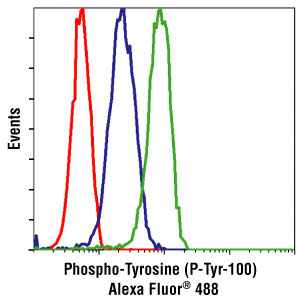
I previously discussed phospho-flow cytometry, a method to study intracellular protein phosphorylation events in peripheral blood mononuclear cells (PBMC) at the single cell level. In standard phospho-flow cytometry protocols, prior to performing assays, PBMCs are first isolated from blood using density gradient centrifugation methods such as Ficoll. However, there may be times when it is advantageous to study signaling pathways in relatively unmanipulated cells directly ex vivo. For this, Chow et al. have established a protocol for performing phospho-flow cytometry on PBMCs directly in whole blood.

There are many advantages to isolating and cryopreserving PBMCs with the intention of later studying signaling events by methods including standard phospho-flow cytometry. In particular, when comparisons are desired between patient groups and healthy controls, there is likely to be less confounding contributions of experimental variability to the results if all of the comparative samples are assayed together. However, as discussed by Chow et al., pharmacodynamic monitoring as well as evaluation of constitutively activated signaling pathways in PBMCs would be best studied on cells having undergone the least manipulation. Some signaling pathway responses may be more robust in whole blood PBMCs as well. For example, I have found in my own assays that signaling responses to IL-6 are strongest in whole blood PBMCs compared with PBMCs following Ficoll or culture in the incubator for any amount of time. This method can also be used to study bone marrow immune cell signaling as well as expression of intracellular molecules that are exposed by the permeabilization method chosen. In addition, looking at signaling events in murine PBMCs is difficult to do if PBMCs need to be isolated first, given the very small amount of blood that can be obtained from a mouse. In these cases, anti-coagulated whole blood phospho-flow cytometry should be considered.
Whole blood phospho-flow cytometry is a relatively easy method. Using 100 ul of whole blood is enough for this assay, and the stimulus (cytokine or other activating signaling molecule) is added directly to the whole blood for the preferred amount of time. PBMCs are then fixed with formaldehyde and a Triton X-100 based buffer is added to lyse the red blood cells and permeabilize the white blood cells. This is followed with a few washes and finally the cells can be treated with methanol to unmask phospho-epitopes, similarly to the standard phospho-flow cytometry method by Nolan and colleagues. Chow et al. include an optional step in which the PBMCs can be stored in a freezing buffer prior to methanol treatment. However, I have successfully stored PBMCs in 90-100% methanol at -20 or -80 ºC until staining for flow cytometry, similarly to what is done for the standard phospho-flow cytometry method by Nolan and colleagues.
As with all protocols involving treatment of cells with reagents such as methanol or Triton X-100, some epitopes may be lost and thus will not be evaluable if staining is done following these treatments. Thus, there is an alternate method included in the protocol to stain for some antigens up front. As a reminder however, some fluorophores are sensitive to methanol, for instance V500, and thus cannot be used to stain PBMCs prior to such treatments. Finally, in a prior article, Chow et al. (2005), tested different methods of fixation, permeabilization and alcohol unmasking, and I have included the link to that article below as an excellent reference in the case that modulation of the protocol is required for optimal assessment of your antigens of interest.
Further Reading:
Whole blood processing for measurement of signaling proteins by flow cytometry. Chow S, Hedley D, Shankey TV. Curr Protoc Cytom. 2008 Oct;Chapter 9:Unit 9.27.
Whole blood fixation and permeabilization protocol with red blood cell lysis for flow cytometry of intracellular phosphorylated epitopes in leukocyte subpopulations. Chow S, Hedley D, Grom P, Magari R, Jacobberger JW, Shankey TV. Cytometry A. 2005 Sep;67(1):4-17.
Single-cell phospho-protein analysis by flow cytometry. Schulz KR, Danna EA, Krutzik PO, Nolan GP.Curr Protoc Immunol. 2012 Feb;Chapter 8:Unit 8.17.1-20.
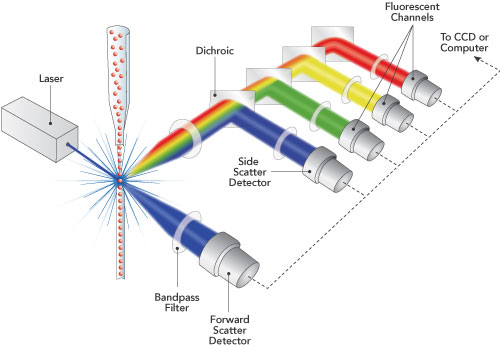


 In
In 
 Gerhard
Gerhard

 In this first half of the two part blog, I will talk about the two reasons, namely unspecific binding and Fc-receptors, which most people think of when they talk about non-specific binding in flow cytometry.
In this first half of the two part blog, I will talk about the two reasons, namely unspecific binding and Fc-receptors, which most people think of when they talk about non-specific binding in flow cytometry.
