

Parkinson’s disease (PD) is a chronic neurodegenerative condition effecting dopaminergic neurons of the midbrain. PD manifests itself around age 50 with mainly motor symptoms, such as tremor (shaking), slowness of movement, rigidity and postural instability. Number of pharmaceutical agents (e.g., L-Dopa and MAO-B inhibitors) has been used for symptomatic relief in PD patients, but the ultimate therapy target is the replacement of degenerating dopaminergic neurons with new, healthy neurons.
 Cell replacement therapy for PD dates back to mid 80s with the transplantation of adrenal medullary tissue into patients’ striatum [1-3], which resulted only moderate improvements. At the same time, researchers in Sweden performed transplantation of fetal ventral mesencephalic tissue from aborted fetuses [4, 5]. These early studies observed important and persistent improvement based on numerous clinical outcomes. Moreover, postmortem examination of the brains of PD patients, who received ventral mesencephalic tissue transplantation, showed sustained survival of the graft and re-innervation of the striatum [6]. With the lift of federal funding ban on using fetal tissue for research and therapy by President Clinton in 1993, United States also began clinical trials utilizing fetal ventral mesencephalic tissue [7, 8]. Unfortunately, not only the patients didn’t display any significant improvements following transplantation in these trials, they developed additional abnormal, involuntary movements (i.e., graft-induced dyskinesia), due to surgery, which was also observed in other trials.
Cell replacement therapy for PD dates back to mid 80s with the transplantation of adrenal medullary tissue into patients’ striatum [1-3], which resulted only moderate improvements. At the same time, researchers in Sweden performed transplantation of fetal ventral mesencephalic tissue from aborted fetuses [4, 5]. These early studies observed important and persistent improvement based on numerous clinical outcomes. Moreover, postmortem examination of the brains of PD patients, who received ventral mesencephalic tissue transplantation, showed sustained survival of the graft and re-innervation of the striatum [6]. With the lift of federal funding ban on using fetal tissue for research and therapy by President Clinton in 1993, United States also began clinical trials utilizing fetal ventral mesencephalic tissue [7, 8]. Unfortunately, not only the patients didn’t display any significant improvements following transplantation in these trials, they developed additional abnormal, involuntary movements (i.e., graft-induced dyskinesia), due to surgery, which was also observed in other trials.
Close examination of the transplantation studies using fetal ventral mesencephalic tissue revealed few noteworthy outcomes:
1. Younger patients with newly developed pathology showed significant improvements over older patients with severe PD pathology.
2. Some patients showed continued improvements 3-4 years after surgery, while they did not display any benefits during the first year, indicating that the improvement in clinical parameters may take a while to appear over time. Regardless, it is clear that patients respond differently to the transplants of dopaminergic neurons, making the clinical outcomes fluctuate considerably.
3. Preparation of the fetal tissues, as well as selection of patients for transplantation, varied significantly from center to center carrying out the clinical trials, further indicating the need for standardizing tissue preparation, patient selection and implantation site.
Compared to the aforementioned points, the use of fetal ventral mesencephalic tissue for grafting constitutes one of the biggest problems in cell based therapy for PD. It has been challenging to standardize the number and the quality of the fetal dopaminergic cells in graft preparations. Furthermore, the purity of the preparations also varies from batch to batch. Lastly, many ethical -and sometimes legal- issues surround fetal tissues/cells significantly limiting their clinical applicability. Do we have an alternative source that is free of these concerns/problems? The answer is yes, but not at the moment. With the isolation of human embryonic stem cells (hESCs) in 1998 and the introduction of human induced pluripotent stem cells (iPSCs) in 2007, stem cell derived dopaminergic neurons are at the top of everyone’s list when it comes to replacing degenerating neurons in PD. hESCs have been the primary source to produce dopaminergic neurons so far [9-11], but with the popularity and the advantages of iPSCs, the focus is more likely to shift to iPSC-derived dopaminergic neurons in future transplantation efforts.
Number of studies utilizing stem cell derived dopaminergic neurons in animal models of PD reported promising results over the years. However, we are far from using these cells in clinical trials. Many issues, such as long-term stability of the transplanted cells, sustained functional recovery, ability to re-innervate the host striatum, generation of GMP grade cells and long-terms safety especially with regards to tumor formation, remain to be determined. To be able to answer these concerns are critical for successful clinical translation of stem cell derived dopaminergic neurons. Nevertheless, the target is in front of everyone, and the field of regenerative medicine is moving at an incredible speed to reach it. It should also be noted that an increasing number of novel therapeutic approaches (e.g., gene therapy and growth factor infusions) have been under development -in addition to cell transplantations- with the aim of restoring dopaminergic function in PD patients.
While we are looking ahead with the promise of stem cell derived dopaminergic neurons for future of cell-based therapy in PD, there are many lessons to be learnt from the early clinical trials using fetal ventral mesencephalic tissue. There is no question that fetal dopamine neurons will serve as a reference and a standard against stem cell derived neurons for future clinical trials, since we know that the transplants survived, re-innervated the striatum, and generated adequate symptomatic relief in some patients for more than a decade following surgery. For PD patients, who are interested in cell-based therapy now, the decision of whether to wait for clinical trials utilizing stem cell derived neurons or to proceed with currently available fetal tissue grafts remains a somewhat difficult question and should take into consideration the aforementioned strengths and weaknesses of each approach.
References:
[1] Backlund EO, Granberg PO, Hamberger B, et al. Transplantation of adrenal medullary tissue to striatum in parkinsonism. First clini- cal trials. J Neurosurg 1985;62:169–173.
[2] Herrera-Marschitz M, Stromberg I, Olsson D, Ungerstedt U, Olson L. Adrenal medullary implants in the dopamine-denervated rat striatum. II. Acute behavior as a function of graft amount and location and its modulation by neuroleptics. Brain Res 1984;297:53–61.
[3] Madrazo I, Drucker-Colin R, Diaz V, Martinez-Mata J, Torres C, Becerril JJ. Open microsurgical autograft of adrenal medulla to the right caudate nucleus in two patients with intractable Parkinson’s disease. N Engl J Med 1987;316:831–834.
[4] Lindvall O, Brundin P, Widner H, et al. Grafts of fetal dopamine neurons survive and improve motor function in Parkinson’s dis- ease. Science 1990;247:574–577.
[5] Widner H, Tetrud J, Rehncrona S, et al. Bilateral fetal mesence- phalic grafting in two patients with parkinsonism induced by 1- methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). N Engl J Med 1992;327:1556–1563.
[6] Kordower JH, Rosenstein JM, Collier TJ, et al. Functional fetal nigral grafts in a patient with Parkinson’s disease: chemoanatomic, ultrastructural, and metabolic studies. J Comp Neurol 1996;370:203–230.
[7] Freed CR, Greene PE, Breeze RE, et al. Transplantation of embry- onic dopamine neurons for severe Parkinson’s disease. N Engl J Med 2001;344:710–719.
[8] Olanow CW, Goetz CG, Kordower JH, et al. A double-blind con- trolled trial of bilateral fetal nigral transplantation in Parkinson’s disease. Ann Neurol 2003;54:403–414.
[9] Lee SH, Lumelsky N, Studer L, Auerbach JM, McKay RD. Effi- cient generation of midbrain and hindbrain neurons from mouse embryonic stem cells. Nat Biotechnol 2000;18:675–679.
[10] Cho MS, Lee YE, Kim JY, et al. Highly efficient and large-scale generation of functional dopamine neurons from human embryonic stem cells. Proc Natl Acad Sci U S A 2008;105:3392–3397.
[11] Kawasaki H, Suemori H, Mizuseki K, et al. Generation of dopami- nergic neurons and pigmented epithelia from primate ES cells by stromal cell-derived inducing activity. Proc Natl Acad Sci U S A 2002;99:1580–1585.
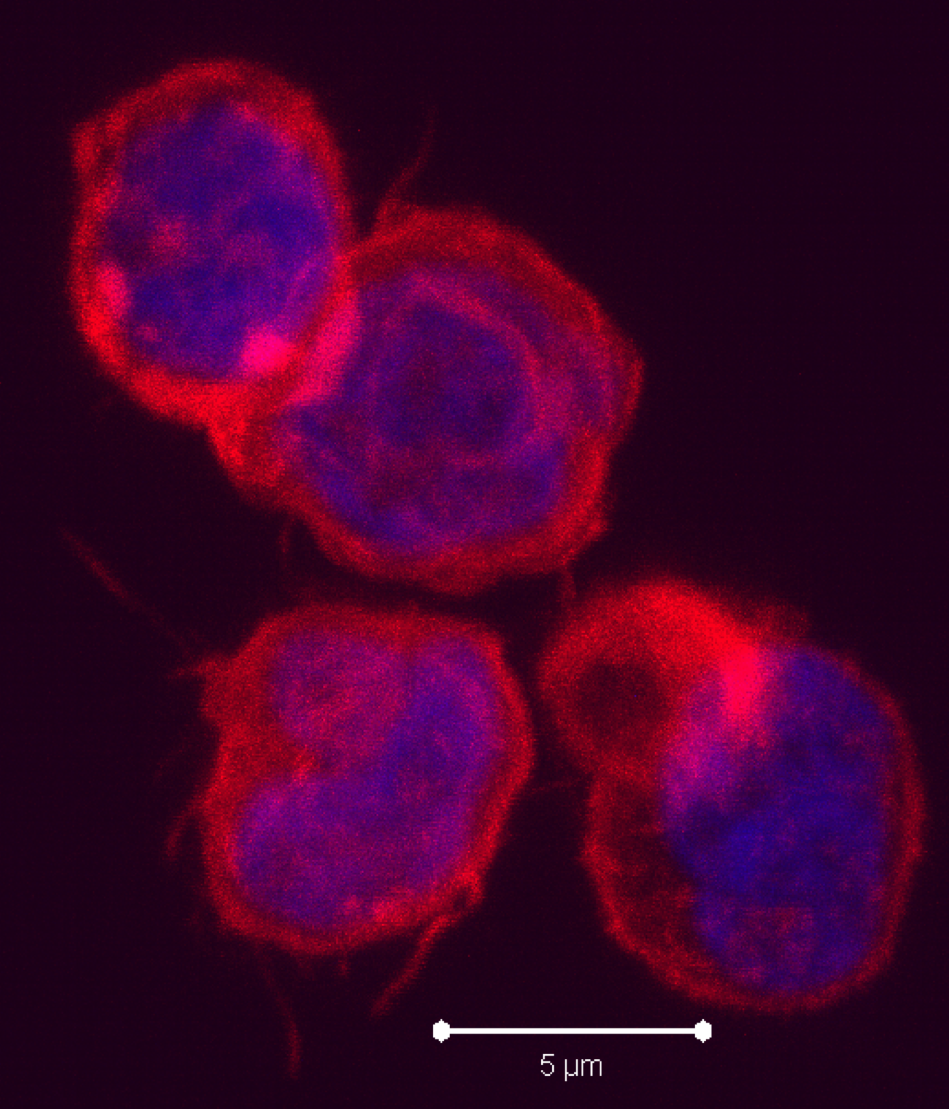
 cells in that they produce IL-5 and IL-13. RORγt+ ILCs aka ILC3s, include subsets that resemble TH17 and TH22 cells by producing IL-17 and IL-22, respectively, as well as a subset that produces both cytokines. Several recent articles have identified another class of ILCs in both humans and mice. These cells resemble TH1 cells in that they express T-bet/TBX21 and produce IFN-gamma, and are distinct from conventional NK cells found among peripheral blood mononuclear cells (PBMC). These newly characterized cellular subsets have been denoted as Type 1 ILCs (ILC1).
cells in that they produce IL-5 and IL-13. RORγt+ ILCs aka ILC3s, include subsets that resemble TH17 and TH22 cells by producing IL-17 and IL-22, respectively, as well as a subset that produces both cytokines. Several recent articles have identified another class of ILCs in both humans and mice. These cells resemble TH1 cells in that they express T-bet/TBX21 and produce IFN-gamma, and are distinct from conventional NK cells found among peripheral blood mononuclear cells (PBMC). These newly characterized cellular subsets have been denoted as Type 1 ILCs (ILC1).

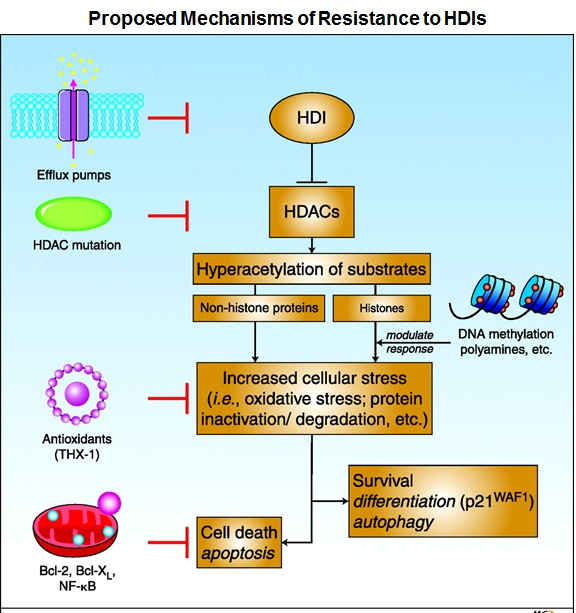
 ion of reactive oxygen species (ROS) scavenger protein thioredoxin, elevated expression of anti-apoptotic proteins Bcl-2 and Bcl-xL, increased expression of histone HDAC enzymes, and activation of several signaling pathways including MAPK, phosphoinositide 3-kinase and signal transducer and activator of transcription.
ion of reactive oxygen species (ROS) scavenger protein thioredoxin, elevated expression of anti-apoptotic proteins Bcl-2 and Bcl-xL, increased expression of histone HDAC enzymes, and activation of several signaling pathways including MAPK, phosphoinositide 3-kinase and signal transducer and activator of transcription.
 At the writing of this blog, no proposed drug has been proposed that directly targets the Hippo signaling pathway. However, many possible targets for therapy are being investigated that would also affect the Hippo signaling pathway
At the writing of this blog, no proposed drug has been proposed that directly targets the Hippo signaling pathway. However, many possible targets for therapy are being investigated that would also affect the Hippo signaling pathway
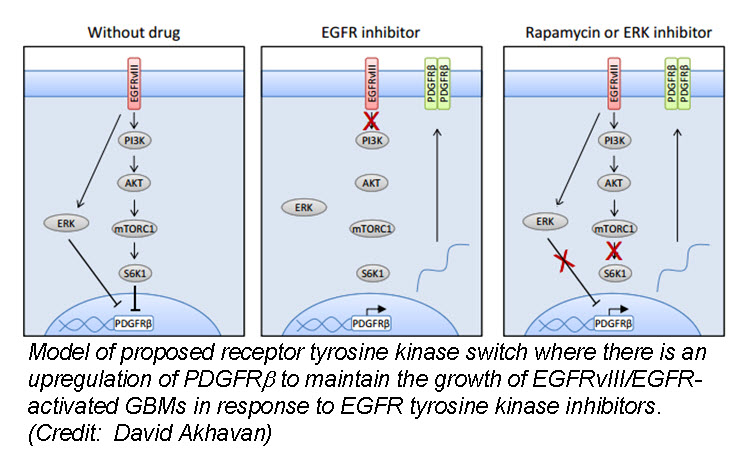
 a (GBM) cells develop resistance to anti-EGFR therapy. They demonstrate for the first time that an EGFR-dependent cancer can escape targeted therapy by developing dependence on another non-amplified, non-mutated RTK. Specifically, they show that GBMs with EGFR mutations evade EGFR tyrosine kinase inhibitors (TKI) by transcriptionally de-repressing platelet-derived growth factor receptor β (
a (GBM) cells develop resistance to anti-EGFR therapy. They demonstrate for the first time that an EGFR-dependent cancer can escape targeted therapy by developing dependence on another non-amplified, non-mutated RTK. Specifically, they show that GBMs with EGFR mutations evade EGFR tyrosine kinase inhibitors (TKI) by transcriptionally de-repressing platelet-derived growth factor receptor β (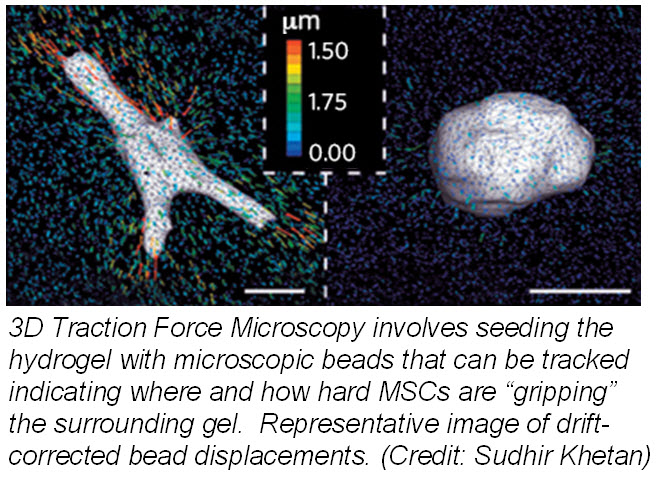
 ranslate that into chemical signals) can influence stem cell fate. Researchers from University of Pennsylvania showed that cell fate is regulated by cell-generated tension that is enabled through cell-mediated degradation of the covalently crosslinked matrix. When cultured on “softer” 2D covalently crosslinked gels (RGD-modified methacrylated hyaluronic acid hydrogels), mesenchymal stem cells (MSCs) differentiated into adipocytes when cultured in bipotential adipogenic/ osteogenic media. In contrast, MSCs cultured on “harder” 2D alginate gels differentiated into chondrocytes. This phenomenon was not present in 3D hydrogels and was attributed to the inability of cells to degrade the covalent cross-linked bonds resulting in MSCs differentiating into adipocytes. Introduction of proteolytically cleavable crosslinks and utilizing
ranslate that into chemical signals) can influence stem cell fate. Researchers from University of Pennsylvania showed that cell fate is regulated by cell-generated tension that is enabled through cell-mediated degradation of the covalently crosslinked matrix. When cultured on “softer” 2D covalently crosslinked gels (RGD-modified methacrylated hyaluronic acid hydrogels), mesenchymal stem cells (MSCs) differentiated into adipocytes when cultured in bipotential adipogenic/ osteogenic media. In contrast, MSCs cultured on “harder” 2D alginate gels differentiated into chondrocytes. This phenomenon was not present in 3D hydrogels and was attributed to the inability of cells to degrade the covalent cross-linked bonds resulting in MSCs differentiating into adipocytes. Introduction of proteolytically cleavable crosslinks and utilizing 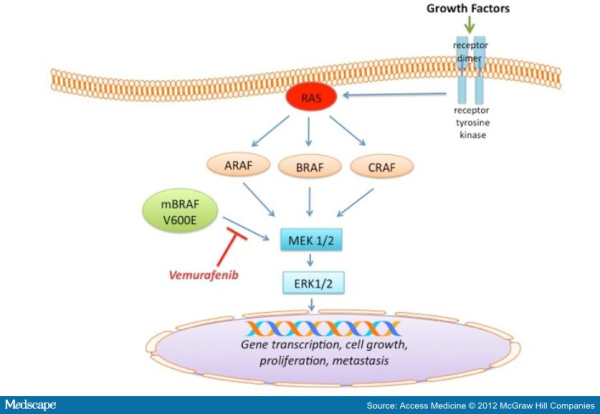
 echanisms of acquired resistance to BRAF inhibitors (for detail please refer to my blog post titled ” RESISTANCE TO B-RAF INHIBITORS IN MELANOMA’’). Due to heterogeneous nature of cancer cells, it is crucial to gain a thorough understanding of the underlying drug-resistance mechanisms so that we can develop novel strategies to circumvent resistance and achieve more-prolonged responses. A recent study published in the journal Nature by Das Thakur and colleagues reported that intermittent treatment of vemurafenib prevented resistance in primary human melanoma xenografts. To study mechanisms of resistance to vemurafenib, Das Thakur et al. developed an animal model by continuously treating mice bearing a vemurafenib-naive, patient-derived BRAF-mutant melanoma with vemurafenib until drug resistance developed. Exome sequence analysis did not detect any secondary mutations in the coding sequences of BRAF, NRAS, KRAS, HRAS, and MEK1 in the resistant tumors. No alternatively spliced isoform of V600EBRAF, another known mechanism of vemurafenib resistance in melanoma was also detected in the resistant tumors. However, increased expression of V600EBRAF protein was noted in the resistant tumors and inhibition of V600EBRAF gene by RNA interference resulted in suppression of proliferation. These data suggested that the tumor cells were BRAF oncogene dependent and the observed drug resistance was due to the increased expression of V600EBRAF protein. In addition to these, another interesting observation was noted in this study when Das Thakur et al. tried to establish cell lines derived from the drug-resistant tumors. Cell lines derived from the drug-resistant tumors could not be developed without vemurafenib, where withdrawal of vemurafenib from the newly established cell lines changed cell morphology and decreased proliferation. This suggested that vemurafenib-resistant tumor cells in melanoma suffer a fitness deficit in the absence of vemurafenib. A similar type of vemurafenib dependency was also observed in SK-MEL239-C3 melanoma cells in which resistance is due to expression of a splice variant of V600EBRAF, and also in tumor cells isolated from a BRAF-mutated vemurafenib-resistant melanoma patient. Consistent with these findings, Das Thakur et al. observed tumor regression within 10 days in mice bearing vemurafenib resistant melanoma following cessation of vemurafenib treatment, although tumors eventually started re-growing. Collectively these results suggested that withdrawal of vemurafenib might create a hostile environment for drug-resistant cells and detain the onset of drug resistance. A comparison study made between continuous and intermittent vemurafenib treatment in human melanoma xenografts bearing mice further validated these observations. Drug resistance was developed in mice with 100 days receiving continuous treatment, whereas none of the mice on the intermittent treatment schedule exhibited drug resistance after 200 days of treatment. Therefore, these findings recommend that discontinuous treatment of vemurafenib may select against drug-resistant cells and prolong the responses to vemurafenib in melanoma. Future studies are needed especially in clinical trials to validate this proposal.
echanisms of acquired resistance to BRAF inhibitors (for detail please refer to my blog post titled ” RESISTANCE TO B-RAF INHIBITORS IN MELANOMA’’). Due to heterogeneous nature of cancer cells, it is crucial to gain a thorough understanding of the underlying drug-resistance mechanisms so that we can develop novel strategies to circumvent resistance and achieve more-prolonged responses. A recent study published in the journal Nature by Das Thakur and colleagues reported that intermittent treatment of vemurafenib prevented resistance in primary human melanoma xenografts. To study mechanisms of resistance to vemurafenib, Das Thakur et al. developed an animal model by continuously treating mice bearing a vemurafenib-naive, patient-derived BRAF-mutant melanoma with vemurafenib until drug resistance developed. Exome sequence analysis did not detect any secondary mutations in the coding sequences of BRAF, NRAS, KRAS, HRAS, and MEK1 in the resistant tumors. No alternatively spliced isoform of V600EBRAF, another known mechanism of vemurafenib resistance in melanoma was also detected in the resistant tumors. However, increased expression of V600EBRAF protein was noted in the resistant tumors and inhibition of V600EBRAF gene by RNA interference resulted in suppression of proliferation. These data suggested that the tumor cells were BRAF oncogene dependent and the observed drug resistance was due to the increased expression of V600EBRAF protein. In addition to these, another interesting observation was noted in this study when Das Thakur et al. tried to establish cell lines derived from the drug-resistant tumors. Cell lines derived from the drug-resistant tumors could not be developed without vemurafenib, where withdrawal of vemurafenib from the newly established cell lines changed cell morphology and decreased proliferation. This suggested that vemurafenib-resistant tumor cells in melanoma suffer a fitness deficit in the absence of vemurafenib. A similar type of vemurafenib dependency was also observed in SK-MEL239-C3 melanoma cells in which resistance is due to expression of a splice variant of V600EBRAF, and also in tumor cells isolated from a BRAF-mutated vemurafenib-resistant melanoma patient. Consistent with these findings, Das Thakur et al. observed tumor regression within 10 days in mice bearing vemurafenib resistant melanoma following cessation of vemurafenib treatment, although tumors eventually started re-growing. Collectively these results suggested that withdrawal of vemurafenib might create a hostile environment for drug-resistant cells and detain the onset of drug resistance. A comparison study made between continuous and intermittent vemurafenib treatment in human melanoma xenografts bearing mice further validated these observations. Drug resistance was developed in mice with 100 days receiving continuous treatment, whereas none of the mice on the intermittent treatment schedule exhibited drug resistance after 200 days of treatment. Therefore, these findings recommend that discontinuous treatment of vemurafenib may select against drug-resistant cells and prolong the responses to vemurafenib in melanoma. Future studies are needed especially in clinical trials to validate this proposal.
 Activation of 41BB on T cells leads to enhanced T cell survival. Anti-41BB-agonistic antibodies have demonstrated significant anti-tumor activity in mice by enhancing anti-tumor cytotoxic T cell responses. Thus, there are currently several clinical trials underway exploring the efficacy of anti-41BB-agonist antibodies in several types of cancers, including melanoma, renal carcinoma, ovarian cancer, and lymphoma. In a previous study by the same group (
Activation of 41BB on T cells leads to enhanced T cell survival. Anti-41BB-agonistic antibodies have demonstrated significant anti-tumor activity in mice by enhancing anti-tumor cytotoxic T cell responses. Thus, there are currently several clinical trials underway exploring the efficacy of anti-41BB-agonist antibodies in several types of cancers, including melanoma, renal carcinoma, ovarian cancer, and lymphoma. In a previous study by the same group (