
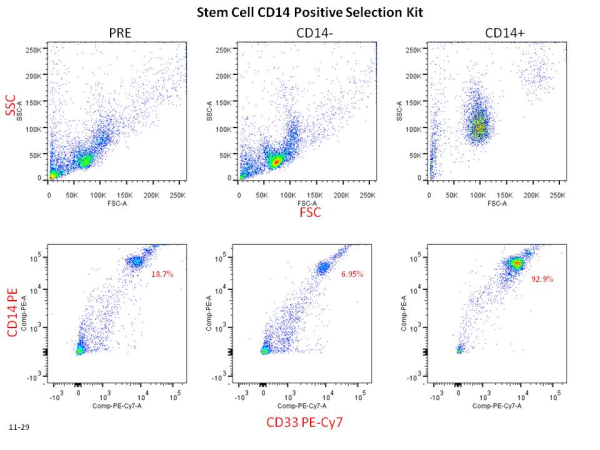
Time of Flight Mass Cytometry (CyTOF) is a relatively new multiparametric technology that is far outpacing standard fluorescence-based flow cytometry in the number of parameters that can be simultaneously assessed on a single cell. In CyTOF, rare transition element isotope-conjugated antibodies are used to label cellular antigens of interest, the magnitude of which is then quantitated by a time of flight mass cytometer, as discussed previously. Previous studies assessing 34 cell surface and intracellular proteins by this technology demonstrated the existence of high dimensional complexity in the heterogeneity of human bone marrow and CD8+ T cell populations. In a July 2013 article in Nature Biotechonology, Newell et al., move CyTOF and the field of immunology another technological step forward by utilizing CyTOF to measure the frequencies of Rotavirus antigen-specific T cells in human peripheral blood mononuclear cells (PBMCs) and jejunal tissue with peptide-MHC tetramers.
In CyTOF, the theoretical maximum number of simultaneously assessable parameters is 100-200 depending on the instrument. This vastly outnumbers the assessable parameters of standard fluorescence-based flow cytometry. To date however, only approximately 40 metal ions have been utilized for antibody labeling, and the development of further metal-chelating technologies is awaited in order to utilize the maximum capacity of the CyTOF instrument. In the current study, the authors circumvent this limitation by using a “bar-coding” methodology in which a variant combination of three out of ten metal ions are used for labeling each tetramer, allowing for up to 120 different metal combinations.
In this study, the authors sought to identify Rotavirus epitopes recognized by human CD8+ T cells in the context of the MHC class I allele, HLA-A*0201. To date, only two Rotavirus epitopes recognized by T cells have been identified, and little is known about the phenotypic and functional diversity of antigen-specific T cells for any particular pathogen. The technical difficulties in proper epitope prediction along with the limited number of cells attainable from human blood samples contribute to these issues. Thus, this method represents a huge leap forward in the potential to identify significantly more antigen-specific T cell epitopes and to extensively classify these cells functionally. Using an MHC-prediction algorithm, 77 possible Rotavirus peptides were identified that bound to HLA-A*0201. An additional 32 positive and negative control tetramers were added for a total of 109 labeled tetramers used to stain each sample simultaneously. This was further combined with 23-27 metal-chelated antibodies specific for cell surface and intracellular antigens to phenotypically characterize the T cells. A specialized Matlab script was used to analyze the high-dimensional data obtained following mass spectrometry of PBMC and jejunal samples.
On average, CD8 T cell populations specific for two Rotavirus-peptides plus 6-7 peptides from other viruses including influenza, EBV, and CMV, were identified on average across PBMCs from the 17 healthy donors analyzed. These antigen-specific T cell populations were further phenotypically characterized by expression of surface and intracellular markers. CD8 T cells specific for six Rotavirus epitopes that included the two previously identified epitopes, were recurrently detected in PBMCs from at least two individuals. Of these, CD8 cells specific for a Rotavirus peptide from the VP3 protein were most common among healthy donor PBMCs and were phenotypically unique, being of the effector memory subtype compared with a central memory phenotype typical of the T cells specific for the other Rotavirus peptides. VP3-specific T cells were also uniquely present in jejunal tissue obtained from obese patients that had undergone gastric bypass surgeries. Thus, this methodology discovered at least 4 new Rotavirus peptides as well as unique characteristics of the different antigen-specific CD8 T cell populations.
In summary, this methodology of combining CyTOF technology with tetramer “bar-coding” paves the way for a vast expansion over fluorescent-based flow cytometry techniques for identifying antigen-specific T cell populations. As vaccine strategies are an ongoing goal for treatment and prevention of infectious diseases and cancer, it is important to not only identify the peptides that can elicit T cell responses, but also functionally characterize these T cells in order to maximally promote desired immune responses.
Further Reading:
Combinatorial tetramer staining and mass cytometry analysis facilitate T-cell epitope mapping and characterization. Newell EW, Sigal N, Nair N, Kidd BA, Greenberg HB, Davis MM. Nat Biotechnol. 2013 Jul;31(7):623-9. doi: 10.1038/nbt.2593. Epub 2013 Jun 9.
Cracking the code of human T-cell immunity. Harvey CJ, Wucherpfennig KW. Nat Biotechnol. 2013 Jul 9;31(7):609-10. doi: 10.1038/nbt.2626.
Cytometry by time-of-flight shows combinatorial cytokine expression and virus-specific cell niches within a continuum of CD8+ T cell phenotypes. Immunity. 2012 Jan 27;36(1):142-52. doi: 10.1016/j.immuni.2012.01.002.
Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Bendall SC, Simonds EF, Qiu P, Amir el-AD, Krutzik PO, Finck R, Bruggner RV, Melamed R, Trejo A, Ornatsky OI, Balderas RS, Plevritis SK, Sachs K, Pe’er D, Tanner SD, Nolan GP. Science. 2011 May 6;332(6030):687-96. doi: 10.1126/science.1198704.




 cells in that they produce IL-5 and IL-13. RORγt+ ILCs aka ILC3s, include subsets that resemble TH17 and TH22 cells by producing IL-17 and IL-22, respectively, as well as a subset that produces both cytokines. Several recent articles have identified another class of ILCs in both humans and mice. These cells resemble TH1 cells in that they express T-bet/TBX21 and produce IFN-gamma, and are distinct from conventional NK cells found among peripheral blood mononuclear cells (PBMC). These newly characterized cellular subsets have been denoted as Type 1 ILCs (ILC1).
cells in that they produce IL-5 and IL-13. RORγt+ ILCs aka ILC3s, include subsets that resemble TH17 and TH22 cells by producing IL-17 and IL-22, respectively, as well as a subset that produces both cytokines. Several recent articles have identified another class of ILCs in both humans and mice. These cells resemble TH1 cells in that they express T-bet/TBX21 and produce IFN-gamma, and are distinct from conventional NK cells found among peripheral blood mononuclear cells (PBMC). These newly characterized cellular subsets have been denoted as Type 1 ILCs (ILC1).
 -presentation is a DC-specialized mechanism by which antigens are taken up through endocytic and phagocytic pathways but presented in the context of MHC-class I, to activate antigen-specific cytotoxic CD8 T cells. Recent studies have sought to characterize the differences between the many tissue-associated DC subsets including their ability to cross-present antigen.
-presentation is a DC-specialized mechanism by which antigens are taken up through endocytic and phagocytic pathways but presented in the context of MHC-class I, to activate antigen-specific cytotoxic CD8 T cells. Recent studies have sought to characterize the differences between the many tissue-associated DC subsets including their ability to cross-present antigen.


 Negative regulatory CD4 T cells are well characterized and highly studied. However their CD8 counterparts are not well defined, particularly in humans. Regulatory CD8 T cells suppress activated CD4 T cells and have proposed roles in various human diseases including multiple sclerosis, ovarian carcinoma and infection with HIV, and many subsets have been described using various markers. In a recent issue of PLoS One, Hu et. al, describe a population of CD3+CD8+CD161−CD56+ T cells within human peripheral blood mononuclear cells (PBMC) that exhibit a cytolytic negative regulatory function.
Negative regulatory CD4 T cells are well characterized and highly studied. However their CD8 counterparts are not well defined, particularly in humans. Regulatory CD8 T cells suppress activated CD4 T cells and have proposed roles in various human diseases including multiple sclerosis, ovarian carcinoma and infection with HIV, and many subsets have been described using various markers. In a recent issue of PLoS One, Hu et. al, describe a population of CD3+CD8+CD161−CD56+ T cells within human peripheral blood mononuclear cells (PBMC) that exhibit a cytolytic negative regulatory function.