

For years, patients and their caregivers were mostly uninformed outsiders as pharmaceutical companies and research institutes developed new drugs and strategies for future growth.
Now, however, in order to increase brand awareness or accelerate predictive medicine development, more and more pharmaceutical and biotechnology companies realize the importance of patient engagement, starting at the pre-clinical R&D process all the way through clinical trials and the commercialization phase.
The Problem:
Despite all the ongoing efforts to engage patients, “Big Pharma ranks near bottom in patient attitudes”. The FiercePharma article from January 18, 2013 briefly describes the outcome of a survey of 500 international, national, and regional patient groups, and their views of the different stages of the drug/device development as well as the paying and distribution process. As expected, high drug pricing was ranked on top of the list as the main cause for negative attitudes towards Big Pharma. The survey also indicates that patients found drug companies “too secretive, doing a poor job of letting patients know about adverse drug event news”. Consequently, many patients have issues with the industry’s integrity.
 Overall, I think companies are slowly getting better in bridging the gap between industry and patients. As we all know, change – whether it needs to occur within a company or the mind of a patient – takes time. Gaining patients’ trust will be a long-term task, an infinite circle of strategizing, implementing, observing, and analyzing. Generally speaking, most companies could still need a healthy portion of empathy. If they really care about patients, they would need to develop a real understanding of patients’ or caregivers’ needs, fears, and reasoning of current attitudes.
Overall, I think companies are slowly getting better in bridging the gap between industry and patients. As we all know, change – whether it needs to occur within a company or the mind of a patient – takes time. Gaining patients’ trust will be a long-term task, an infinite circle of strategizing, implementing, observing, and analyzing. Generally speaking, most companies could still need a healthy portion of empathy. If they really care about patients, they would need to develop a real understanding of patients’ or caregivers’ needs, fears, and reasoning of current attitudes.
The good news is: There are several ways to connect with patients on a deeper level.
Depending on a) purpose, b) stage of drug or biomarker development, and c) patient or, eventually, caregiver demographics, one has to develop appropriate recruitment/engagement strategies.
Below, I am outlining some of my thoughts that are based on my own experiences as well as reading many articles and blog posts. It is not a complete list of current status/issues/solutions, and is written to rather start a discussion.
Nevertheless, here are my 2 cents about current developments:
A) Pre-clinical Drug Development
The status quo:
For drug target/validation studies or biomarker development researchers oftentimes need larger quantities of patient samples (often, from hundreds of patients) in order to obtain statistically significant data. Hospital-based biobanks and, eventually, independent biobanks, are organizations that usually have direct access to patients. Although some US-based hospitals have established patient-friendly/engaging websites, those are not used to source samples “on demand”. As a result, it becomes quite time-consuming to source needed samples to start or continue certain research projects.
larger quantities of patient samples (often, from hundreds of patients) in order to obtain statistically significant data. Hospital-based biobanks and, eventually, independent biobanks, are organizations that usually have direct access to patients. Although some US-based hospitals have established patient-friendly/engaging websites, those are not used to source samples “on demand”. As a result, it becomes quite time-consuming to source needed samples to start or continue certain research projects.
The Problem:
As Matt Jones, staff reporter for GenomeWeb Daily News, pointed out in his most recent article:
- Two-thirds of the nation’s biobanks were established over the past decade – it is estimated that there are about 800 biobanks (with 90% embedded within institutions) within the US.
- The increase of genomics and related large-scale studies have led biobanks to play an increasingly central part in biomedical R&D – genomic research appears as if it is a key driver in the “biobank explosion”.
In short, based on increasing demand, competition is increasing, and given the fact that sequencing costs are rapidly decreasing, one can imagine that this trend will continue. But as biobanks increase and specific patient populations stay the same (especially rare diseases), sample/data sourcing becomes a big issue.
Here is another issue to think about:
Matt Jones also wrote: “Several issues that have been flagged by the National Human Genome Research Institute’s Ethical, Legal, and Social Implications Research program are stirred up by the expansion of biobanks, such as questions about policies governing data sharing and security, privacy and the identifiability of genomic information, how and when to return research results and incidental findings, how governance structures function at genomic repositories, and informed consent issues caused by the multiple uses for samples by genome researchers.”
In short, with all the data being generated, certain new regulations might “kick in” within the next few years. Privacy protection always was, and increasingly will be, a topic of many future discussions – especially after the Yaniv Ehrlich’s recent Science article: “Identifying Personal Genomes by Surname Inference”. No matter what your attitude (as a researcher) towards these findings is, patients that become aware of these potential “threats” to privacy will increasingly fear breach of “protected” private data and therefore probably be less willing to donate and share their valuable biosamples and medical information. (If interested, further comments can be read in a New York Times article from January 17, 2013: “Web Hunt for DNA Sequences Leaves Privacy Compromised”).
Why engage with patients?
 It is necessary to connect and build trust. Real trust, however, can only be established through an open, genuine 2-way communication. Since there are already many articles written about this topic, I will not go too far into any details. However, patients/caregivers could potentially be engaged through social media (despite IRB regulations and/or oftentimes long turnaround times) and/or patient portals: for instance, at the biobank Sanguine Biosciences’ patient blog, patients are writing about their experiences, their fears and how they overcame those. At Sanguine, we realize that it is not only important to educate (potential) customers (as Sanguine does too: here is our researcher blog) but even more important to actually work with patients to learn about and educate them – learning, listening and educating needs to go both ways.
It is necessary to connect and build trust. Real trust, however, can only be established through an open, genuine 2-way communication. Since there are already many articles written about this topic, I will not go too far into any details. However, patients/caregivers could potentially be engaged through social media (despite IRB regulations and/or oftentimes long turnaround times) and/or patient portals: for instance, at the biobank Sanguine Biosciences’ patient blog, patients are writing about their experiences, their fears and how they overcame those. At Sanguine, we realize that it is not only important to educate (potential) customers (as Sanguine does too: here is our researcher blog) but even more important to actually work with patients to learn about and educate them – learning, listening and educating needs to go both ways.
Furthermore, increased transparency is another way to develop trust. In the near future, Sanguine Biosciences will notify patients about the impact their samples made by informing them about where they have been sent to and/or for what purpose they were used for. For instance: “Today, we sent some of your serum and CSF to a pharmaceutical company located on the East Coast. This company is seeking to develop a new biomarker that will help detect Alzheimer’s disease at a very early stage. We thank you for helping us accelerate research for Alzheimer’s!”
Sure, some companies or research institutes will not allow us to communicate any information at all; however, others are very interested in working with us to help them bridge the company-patient-gap.
Additional engagement strategies include partnerships/collaborations with non-profit organizations to provide patients the right education and to learn first-hand about the difficulties patients are dealing with on a daily basis. Engaged patients are more likely to share their experiences with others. This leads to a word-of-mouth campaign, which can help increase participation in research studies.
Ultimately, if there is already an existing hospital/biobank-patients-bond, sourcing samples and data will be faster, and less costly, and longitudinal collection studies will become easier. Recruiting donors for clinical trials (i.e. based on their genetic profile) will become easier too.
B) Clinical Trials Phase
 Oftentimes, pharmas, CROs, clinics/hospitals are faced with relatively high patient recruitment costs (about $2000/patient) and/or lengthy recruitment periods. Having engaged patients already “on-hand” might therefore lead to reduced recruitment cost and time. Besides, and as mentioned above, a 2-way dialogue (as much as possible) will help companies build better reputations.
Oftentimes, pharmas, CROs, clinics/hospitals are faced with relatively high patient recruitment costs (about $2000/patient) and/or lengthy recruitment periods. Having engaged patients already “on-hand” might therefore lead to reduced recruitment cost and time. Besides, and as mentioned above, a 2-way dialogue (as much as possible) will help companies build better reputations.
The Pfizer-way:
As a former Pfizer R&D executive LaMattina suggested, big pharma should consider providing more transparency regarding payments to physicians and data derived from clinical trials. In addition, LaMattina advised “to stop pushing drugs for unapproved uses and give up television advertising”. Despite some strong criticism, several pharmaceutical companies already started following these recommendations.
However, regulations will always be a barrier for an open 2-way dialogue. As Todd Kolm, Director, Emerging Channel Strategy at Pfizer pointed out: “Because of the regulatory environment, we’re not able to engage in a true dialogue”.
Furthermore, what makes patient engagement even more difficult is the relatively poor understanding of new media channels, a lack of FDA guidance, a lack of pharmaceutical executives promoting it, the scarcity of demonstrated success stories, the often inadequate resources allocated to new patient engagement strategies, and the simple fact that it is often not really perceived as a must-have, value-added strategy.
Nonetheless, Pfizer’s REMOTE (Research on Electronic Monitoring of OAB (overactive bladder) Treatment Experience) trial proved that through innovative thinking, courage and implementation of completely new strategies, hurdles can slowly be overcome.
REMOTE, which ended in mid-2012, was the first randomized virtual clinical trial conducted under an IND application.
The main problem, however, was recruiting the right patients needed for the study. This was mainly caused by being very regulatory-cautious so no wrong patients would enter the virtual trial. Ultimately, it became very difficult for patients to actually get into the study.
To quote Craig Lipset, Worldwide Head of Clinical Innovation at Pfizer: “As an industry, we will continue to fail to recruit patients in our studies if we cannot create an ecosystem of patients already engaged and aware about research studies and research participation”.
The good news: Having a web-based recruiting site simplified patient recruitment (including consenting) and allowed patients to participate from their own home – web-based and mobile platforms were used to capture self-reported data from those patients. In addition, primary care physicians engaged with patients by screening and caring for them during the trial. Furthermore, patients received their data at the end of the trial period and were able to even share those with their physician.
C) Post-approval/Commercialization Phase
Pharmaceutical biotechnology companies are slowly becoming more patient-centric. Belgium-based UCB, for example, developed a very patient-focused website, engaging patients through education and disease-specific patient-support programs.
Other companies are also trying to bridge the gap to their patients. Major firms such as Biogen Idec , Pfizer, and French pharmaceutical giant Sanofi, the parent of Genzyme, are partnering on a number of initiatives with patient groups such as The Michael J. Fox Foundation for Parkinson’s Research.
Taken together, it is key to provide top-quality, objective, reliable, unbiased education, training and information to patient organizations and patients at large on all aspects of R&D, in order for patients to get involved and become empowered players in the medical drug development process.
Companies that stay on top of this and develop new strategies will ultimately create the necessary paradigm shift, making sure that patients’ needs and insights are at the center in all relevant areas of medical R&D.
Further Reading:
Big Pharma ranks near bottom in patient attitudes – FiercePharma http://www.fiercepharma.com/story/big-pharma-ranks-near-bottom-patient-attitudes/2013-01-18#ixzz2JEbcBxOV
US Sees Boom in Diverse Range of Biobanks, But Regulations are Lacking – GEN http://www.genomeweb.com/us-sees-boom-diverse-range-biobanks-regulations-are-lacking
Identifying Personal Genomes by Surname Inference – Science http://www.sciencemag.org/content/339/6117/321.abstract
Web Hunt for DNA Sequences Leaves Privacy Compromised – New York Times http://www.nytimes.com/2013/01/18/health/search-of-dna-sequences-reveals-full-identities.html?pagewanted=1&_r=0
Between patients and pharmas online, a disconnect – Medical Marketing & Media http://www.mmm-online.com/between-patients-and-pharmas-online-a-disconnect/article/260723/
Pfizer Perseveres In Pioneering Virtual Clinical Trials – Life Science Leader http://lifescienceleader.com/blogs/contributing-editors-2/item/4363-pfizer-perseveres-in-pioneering-virtual-clinical-trials
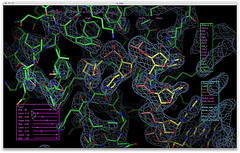
 p53 (TP53), a transcription factor activated by various types of cellular stress, is one of the most well studied tumor suppressor proteins. Stress signals such as various types of DNA damage lead to p53 activation and subsequent induction of genes involved in cell cycle arrest, DNA repair, and apoptosis.
p53 (TP53), a transcription factor activated by various types of cellular stress, is one of the most well studied tumor suppressor proteins. Stress signals such as various types of DNA damage lead to p53 activation and subsequent induction of genes involved in cell cycle arrest, DNA repair, and apoptosis.

 536-47, Figure 3.jpg)
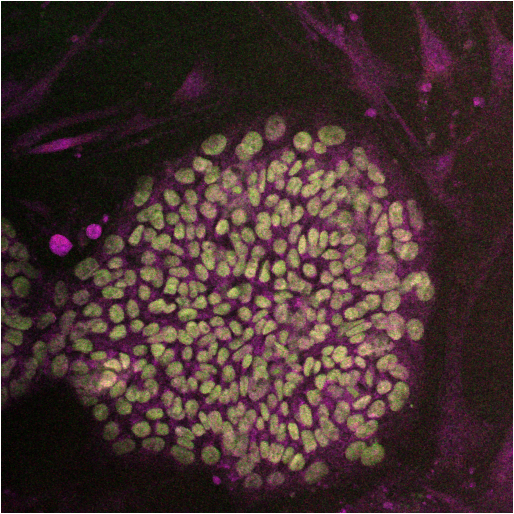


 Blood is considered to be a connective tissue both functionally and embryologically. It originates from the mesodermal layer, the same germ layer that gives rise to the other connective tissues such as bone, cartilage and muscle. Blood cells and blood vessels develop in parallel and form a functional circulatory system. Various studies have shown that hematopoietic differentiation of PSCs in vitro closely resembles early steps of blood development in the embryo and induces blood forming cell populations with mesodermal and hemato-endothelial properties [1]. Different types of mature blood cells were successfully generated from murine, primate and human pluripotent stem cells. Here, we will briefly review the major in vitro systems of hematopoietic differentiation from PSCs.
Blood is considered to be a connective tissue both functionally and embryologically. It originates from the mesodermal layer, the same germ layer that gives rise to the other connective tissues such as bone, cartilage and muscle. Blood cells and blood vessels develop in parallel and form a functional circulatory system. Various studies have shown that hematopoietic differentiation of PSCs in vitro closely resembles early steps of blood development in the embryo and induces blood forming cell populations with mesodermal and hemato-endothelial properties [1]. Different types of mature blood cells were successfully generated from murine, primate and human pluripotent stem cells. Here, we will briefly review the major in vitro systems of hematopoietic differentiation from PSCs. pluripotent stem cells grown in non-adherent culture conditions (3D system). Differentiation of PSCs in aggregates mimics three-dimensional embryonic development and yields the establishment of cell adhesion, paracrine signaling and a microenvironment similar to native tissue structures. Thus, EB formation is often used as a method for initiating spontaneous differentiation of PSCs towards all three germ lines.
pluripotent stem cells grown in non-adherent culture conditions (3D system). Differentiation of PSCs in aggregates mimics three-dimensional embryonic development and yields the establishment of cell adhesion, paracrine signaling and a microenvironment similar to native tissue structures. Thus, EB formation is often used as a method for initiating spontaneous differentiation of PSCs towards all three germ lines.
 histone tails. HDACs also cause deacetylation of non-histone proteins thus altering the transcriptional activity of p53 (tumor suppressor gene), E2F (transcription factor), c-Myc (transcription factor), nuclear factor kB (NF-kB), hypoxia inducible factor 1α (HIF-1 α), estrogen receptor α, and androgen receptor complexes.
histone tails. HDACs also cause deacetylation of non-histone proteins thus altering the transcriptional activity of p53 (tumor suppressor gene), E2F (transcription factor), c-Myc (transcription factor), nuclear factor kB (NF-kB), hypoxia inducible factor 1α (HIF-1 α), estrogen receptor α, and androgen receptor complexes. currently in clinical trials both in monotherapy and in combination therapy with other anti-tumor drugs. A review by Tan et al. (2010) reported that at least 80 clinical trials are underway, testing more than 11 different HDIs in hematologic and solid tumors, including leukemias, lymphomas, and multiple myeloma, lung, breast, pancreas, renal, and bladder cancers, melanoma, glioblastoma. To date, most of the responses using HDIs as single agents were observed in advanced hematologic tumors and few were observed in solid tumors. In 2006, HDI vorinostat (suberoylanilide hydroxamic acid, SAHA) was approved by the Food and Drug Administration (FDA, USA) for the treatment of relapsed and refractory cutaneous T-cell lymphoma CTCL. In November, 2009, the FDA also approved another HDI romidepsin (depsipeptide) for the treatment of CTCL, and in 2011 for the treatment of peripheral T-cell lymphoma patients who have already received prior therapy.
currently in clinical trials both in monotherapy and in combination therapy with other anti-tumor drugs. A review by Tan et al. (2010) reported that at least 80 clinical trials are underway, testing more than 11 different HDIs in hematologic and solid tumors, including leukemias, lymphomas, and multiple myeloma, lung, breast, pancreas, renal, and bladder cancers, melanoma, glioblastoma. To date, most of the responses using HDIs as single agents were observed in advanced hematologic tumors and few were observed in solid tumors. In 2006, HDI vorinostat (suberoylanilide hydroxamic acid, SAHA) was approved by the Food and Drug Administration (FDA, USA) for the treatment of relapsed and refractory cutaneous T-cell lymphoma CTCL. In November, 2009, the FDA also approved another HDI romidepsin (depsipeptide) for the treatment of CTCL, and in 2011 for the treatment of peripheral T-cell lymphoma patients who have already received prior therapy.


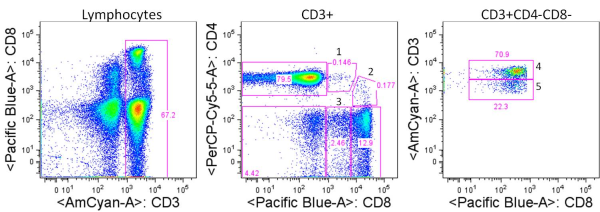
 Significant steps forward are being made in immunotherapeutic approaches for treatment of cancer. Over the past few years, two cancer immunotherapeutics were FDA approved. In 2010, Dendreon Corporation’s Provenge, an autologous cellular vaccine, was approved for hormone refractory metastatic prostate cancer. In 2011, Bristol-Myers Squibb’s anti-CTLA4 antibody Ipilimumab, was FDA approved for late stage melanoma. Recent promising clinical trial results indicate several additional immune modulating therapies are likely to join this prestigious list in the coming years. In addition, combinations of immune therapies with chemotherapeutics are being tested in clinical trials. In light of this, it is important to know how chemotherapies interact with the immune system, in order to best generate synergistic effects.
Significant steps forward are being made in immunotherapeutic approaches for treatment of cancer. Over the past few years, two cancer immunotherapeutics were FDA approved. In 2010, Dendreon Corporation’s Provenge, an autologous cellular vaccine, was approved for hormone refractory metastatic prostate cancer. In 2011, Bristol-Myers Squibb’s anti-CTLA4 antibody Ipilimumab, was FDA approved for late stage melanoma. Recent promising clinical trial results indicate several additional immune modulating therapies are likely to join this prestigious list in the coming years. In addition, combinations of immune therapies with chemotherapeutics are being tested in clinical trials. In light of this, it is important to know how chemotherapies interact with the immune system, in order to best generate synergistic effects.