Patients with autoimmune rheumatic diseases (ARD) such as rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE) have a significantly increased risk of developing cardiovascular disease (CVD) and often develop CVD earlier than those without underlying autoimmunity, although it is not clear whether CVD is a general consequence of RA and SLE or only affects a subgroup of patients. Control of autoimmune inflammation by disease-modifying anti-rheumatic drugs (DMARD), especially those that target immune factors also involved in vasculitis (e.g., T and B cells), is believed to have a protective effect. One area of current research is focused on identifying commonalities across multiple ARD that suggest specific mechanisms of ARD-related CVD in order to develop diagnostics, preventatives, and treatments for those at greatest risk.

A recent article in the Journal of Autoimmunity suggests anti-progranulin antibodies as one potential mechanism. Thurner and colleagues used a protein macro-array to screen serum from patients with anti-neutrophil cytoplasmic antibody (ANCA)-associated systemic vasculitides for novel autoantibodies specific to these diseases. Of the six candidate autoantigens reactive with pooled vasculitis patient serum, progranulin was the only autoantigen appearing in every one of the vasculitides studied. However, extended screenings showed that a positive progranulin antibody titer was not specific for vasculitides; although the prevalence was low in healthy controls (1/97 or 1%) and patients with melanoma (0/98) or sepsis (0/22), progranulin antibodies were also detectedin serum from patients with RA (16/44 or 36%) and SLE (39/91 or 43%).
Progranulin, also called proepithelin, granulin-epithelin precursor, or acrogranin, is a glycoprotein secreted by epithelial cells, neurons, and certain leukocytes. In addition to growth factor-like activity, progranulin has immunomodulatory effects in vitro and in vivo. Full-length progranulin decreases oxidant production by activated neutrophils, blocks TNFα-induced immune responses via binding to TNFR-1 and -2, and promotes up-regulation of IL-4, IL-5, and IL-10. Progranulin deficiency in mice results in greater inflammation in collagen-induced arthritis (CIA) and collagen antibody-induced arthritis models of human RA; treatment of either progranulin-deficient or wild-type mice with recombinant human progranulin ameliorates CIA inflammation.
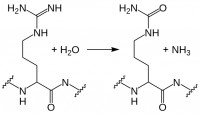
Progranulin is cleaved by several proteases into mature granulins. Neither recombinant nor proteolytically released granulins antagonize TNFα. Rather, granulins increase expression of pro-inflammatory cytokines IL-1β, IL-8, and TNFα. SLPI and apolipoprotein A-I binding to progranulin protects it from cleavage by matrix metalloproteinases and other proteases. However, during inflammation, neutrophils and macrophages release serine proteases that increase progranulin digestion. In the context of ongoing inflammation in ARD, this may result in increased cleavage of anti-inflammatory progranulin to pro-inflammatory granulin.
Thurner et al. are the first to report the presence of neutralizing anti-progranulin antibodies in RA, SLE, and small- and medium-vessel vasculitides, which may represent a pro-inflammatory mechanism common to several autoimmune diseases. Their findings provide additional support for exploration of the progranulin/granulin pathway as a therapeutic target and suggest the potential use of anti-progranulin antibodies as a diagnostic and/or prognostic tool in ARD. Further studies using sera of patients with known autoimmune disease states are needed to confirm these findings and address the additional questions raised, such as – What causes the failure of self-tolerance to progranulin and the generation of anti-progranulin antibodies, as seen in ~20-40% of the patients in this study? Are these anti-progranulin antibodies common to all autoimmune diseases? Could the development of progranulin-neutralizing antibodies even become a biomarker in ARD, for example as a predictor of responsiveness to DMARD therapy, or an indicator of future progression to ARD-related CVD? We await the results of these and other studies in this area with great interest.
Further Reading:
Progranulin antibodies in autoimmune diseases. Thurner L, Preuss KD, Fadle N, Regitz E, Klemm P, Zaks M, Kemele M, Hasenfus A, Csernok E, Gross WL, Pasquali JL, Martin T, Bohle RM, Pfreundschuh M. J Autoimmun. 2013 May; 42:29-38.
Insights into the role of progranulin in immunity, infection, and inflammation. Jian J, Konopka J, Liu C. J Leukoc Biol. 2013 Feb; 93(2):199-208.
Cardiovascular disease in autoimmune rheumatic diseases. Hollan I, Meroni PL, Ahearn JM, Cohen Tervaert JW, Curran S, Goodyear CS, Hestad KA, Kahaleh B, Riggio M, Shields K, Wasko MC. Autoimmun Rev. 2013 Aug; 12(10):1004–1015.