
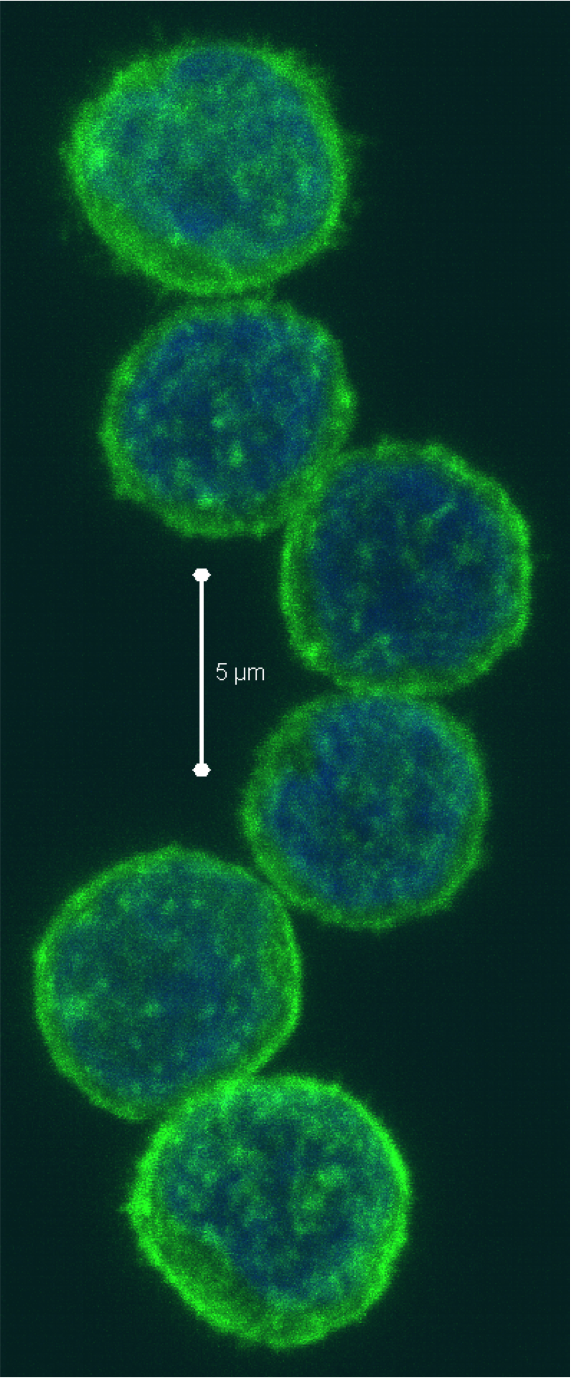
 The availability of human peripheral blood mononuclear cells (PBMC) from healthy individuals and from patients with various diseases allows for many studies on normal and abnormal functions of human immune cells. Because human and murine immune biology differs in many ways, it is important that various methodologies for studying human immunology are established. The two reports highlighted below demonstrate the usage of human PBMCs for mechanistic and pre-clinical human immune cell studies.
The availability of human peripheral blood mononuclear cells (PBMC) from healthy individuals and from patients with various diseases allows for many studies on normal and abnormal functions of human immune cells. Because human and murine immune biology differs in many ways, it is important that various methodologies for studying human immunology are established. The two reports highlighted below demonstrate the usage of human PBMCs for mechanistic and pre-clinical human immune cell studies.
A recent report in The Journal of Immunology by Edwards et. al, demonstrated the usage of healthy human PBMC to elucidate the mechanisms involved in modulation of TH2 T cell responses by Toll-Like Receptor-7 (TLR7) agonists. Because TLR7 stimulation perturbs TH2 responses, the potential use of rapidly metabolized 8-oxoadenine TLR7 antedrugs for treatment of allergic diseases was explored in this study in collaboration between AstraZeneca and Dainippon Sumitomo. The TLR7 agonistic antedrug AZ12441970 was found to inhibit TH2 responses via at least two different mechanisms: TLR7-induced production of type I interferons (IFN) and induction of Notch-ligand expression on TLR7-responsive antigen presenting cells. These led to inhibition of IL-5 production by T cells via the IFN and Notch signaling pathways, respectively. TLR7-induction of IFNα was found to be intact in PBMCs from asthmatic patients when compared with healthy volunteers, and thus the authors proposed that this therapeutic strategy may be effective in allergic disease patients.
This study provides a demonstration of the usage of human PBMCs for elucidating signaling and immune-cell crosstalk mechanisms as well as determining the potential for the effectiveness of candidate drugs in patients with different disease states.
TLR7 Stimulation of APCs Results in Inhibition of IL-5 through Type I IFN and Notch Signaling Pathways in Human Peripheral Blood Mononuclear Cells. Edwards S, Jones C, Leishman AJ, Young BW, Matsui H, Tomizawa H, Murray CM, Biffen M. J Immunol. 2013 Mar 15;190(6):2585-92. doi: 10.4049/jimmunol.1200780.
The role of the cytokine IL-17 in cancer immunity continues to be controversial. IL-17 and the IL-17-producing TH17 T cell subsets have been shown to have both pro-tumor as well as anti-tumor immune modulating functions in different cancer contexts. Monocytes isolated from human PBMC can be differentiated into various myeloid cell types including dendritic cells (DC), providing a tool for studies on these human immune cell types. In a recent Plos One article, Olsson Åkefeldt et. al, explore the role of IL-17 in survival of human monocyte-derived DCs in vitro, and the relevance of this during chemotherapy.
IL-17 was found to significantly prolong DC survival in vitro, however the cells took on expression of macrophage markers (CD14/CD68) and exhibited a pre-M2 macrophage phenotype. The prolonged survival was associated with upregulated expression of pro-survival gene BCL-2A1. Interestingly, IL-17 plus IFNγ treatment in vitro rendered these M2 macrophage-like DCs resistant to cell death induced by 11 of 17 tested chemotherapeutic agents. Thus, to determine if IL-17 treatment would benefit patients by allowing DC survival during therapy, future studies should address whether this chemoresistance of IL-17 treated DCs occurs in patients undergoing chemotherapy, and to determine how IL-17 affects the anti- versus pro-tumor function of these DCs in various types of cancer.
Chemoresistance of Human Monocyte-Derived Dendritic Cells Is Regulated by IL-17A. Olsson Åkefeldt S, Maisse C, Belot A, Mazzorana M, Salvatore G, Bissay N, Jurdic P, Aricò M, Rabourdin-Combe C, Henter JI, Delprat C. PLoS One. 2013;8(2):e56865. doi: 10.1371/journal.pone.0056865. Epub 2013 Feb 18.
Studies like these are examples of the utility of using human PBMCs to elucidate mechanisms of human immune cell biology under normal and diseased conditions.
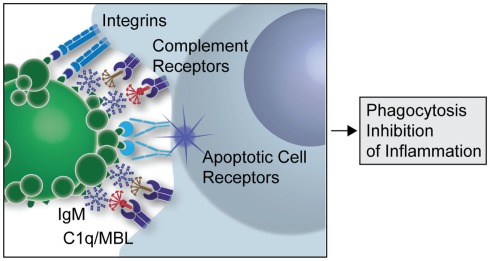
 Figure 1: A simplified overview of the clearance of apoptotic cells by natural IgMs via recruitment of complement factors (Silverman 2012)
Figure 1: A simplified overview of the clearance of apoptotic cells by natural IgMs via recruitment of complement factors (Silverman 2012)
 Arijit
Arijit




 In
In  Gerhard
Gerhard
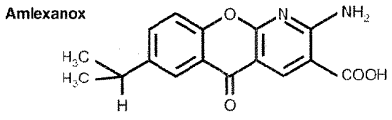
 Can amlexanox, a small molecule drug that has been approved for asthma, allergic rhinitis and aphthous ulcers be an effective treatment for obesity? Many are excited about this prospective presented in a recent article in Nature Medicine by Reilly et. al. as simply Googling “amlexanox” and “obesity” resulted in a plethora of news articles on this report. Interestingly, the two highly related proteins allegedly inhibited by amlexanox that led to this result in mice are the IRF3-kinases, TBK1 (TANK-binding Kinase-1, T2K, NAK) and IKK-ε (IκB kinase-epsilon, IKK-i), whose major known functions are during pathogen infections: the activation of IRF-3 (interferon regulatory kinase-3), the major transcription factor regulating expression of interferon-β (IFNβ).
Can amlexanox, a small molecule drug that has been approved for asthma, allergic rhinitis and aphthous ulcers be an effective treatment for obesity? Many are excited about this prospective presented in a recent article in Nature Medicine by Reilly et. al. as simply Googling “amlexanox” and “obesity” resulted in a plethora of news articles on this report. Interestingly, the two highly related proteins allegedly inhibited by amlexanox that led to this result in mice are the IRF3-kinases, TBK1 (TANK-binding Kinase-1, T2K, NAK) and IKK-ε (IκB kinase-epsilon, IKK-i), whose major known functions are during pathogen infections: the activation of IRF-3 (interferon regulatory kinase-3), the major transcription factor regulating expression of interferon-β (IFNβ).
 CXCR5 is a chemokine receptor expressed by and used to identity human CD4+ follicular helper T cells (TFH). TFH cells, as their name implies, promote the differentiation and survival of memory and plasma B cells in the B cell follicular and germinal center regions of secondary lymphoid organs. CXCR5+ TFH-like central memory CD4+ T cells (CD4+ TCM) also circulate in peripheral blood and can be detected among human peripheral blood mononuclear cells (PBMC). CXCR5+ cells comprise 20-25% of CD4+ TCM cells in human PBMC. However, what are the identifying markers and functional differences of CXCR5+ vs. CXCR5– CD4+ T cells from human PBMC and the prototypical CXCR5+ TFH cells in secondary lymphoid organs?
CXCR5 is a chemokine receptor expressed by and used to identity human CD4+ follicular helper T cells (TFH). TFH cells, as their name implies, promote the differentiation and survival of memory and plasma B cells in the B cell follicular and germinal center regions of secondary lymphoid organs. CXCR5+ TFH-like central memory CD4+ T cells (CD4+ TCM) also circulate in peripheral blood and can be detected among human peripheral blood mononuclear cells (PBMC). CXCR5+ cells comprise 20-25% of CD4+ TCM cells in human PBMC. However, what are the identifying markers and functional differences of CXCR5+ vs. CXCR5– CD4+ T cells from human PBMC and the prototypical CXCR5+ TFH cells in secondary lymphoid organs?
 In this first half of the two part blog, I will talk about the two reasons, namely unspecific binding and Fc-receptors, which most people think of when they talk about non-specific binding in flow cytometry.
In this first half of the two part blog, I will talk about the two reasons, namely unspecific binding and Fc-receptors, which most people think of when they talk about non-specific binding in flow cytometry.



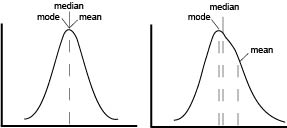
 The first point of confusion is born from the name itself. MFI is often used without explanation, to abbreviate either arithmetic mean, geometric mean, or median fluorescence intensity. In a perfect world, our data would be normally distributed and in that case means, median and mode are all equal. In reality, flow data is rarely normal and never perfect. The more that the data skews, the further the mean drifts in the direction of skew and becomes less representative of the data being analyze as seen on the graphical representation.
The first point of confusion is born from the name itself. MFI is often used without explanation, to abbreviate either arithmetic mean, geometric mean, or median fluorescence intensity. In a perfect world, our data would be normally distributed and in that case means, median and mode are all equal. In reality, flow data is rarely normal and never perfect. The more that the data skews, the further the mean drifts in the direction of skew and becomes less representative of the data being analyze as seen on the graphical representation. Adam
Adam
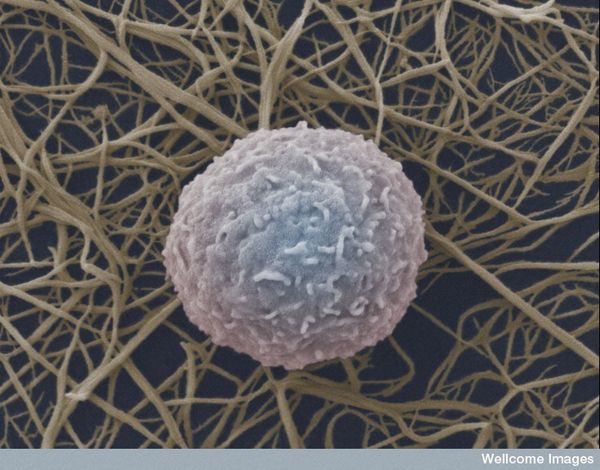
 Natural Killer (NK) cells are a cytotoxic innate immune lymphocyte cell type. In humans, NK cells comprise up to 15% of peripheral blood mononuclear cells (PBMC), and 5-20% of the PBMC lymphocyte population. Several subtypes of NK cells exist in humans. In this post, I will discuss phenotypic properties and markers of NK subtypes present in human PBMC.
Natural Killer (NK) cells are a cytotoxic innate immune lymphocyte cell type. In humans, NK cells comprise up to 15% of peripheral blood mononuclear cells (PBMC), and 5-20% of the PBMC lymphocyte population. Several subtypes of NK cells exist in humans. In this post, I will discuss phenotypic properties and markers of NK subtypes present in human PBMC.