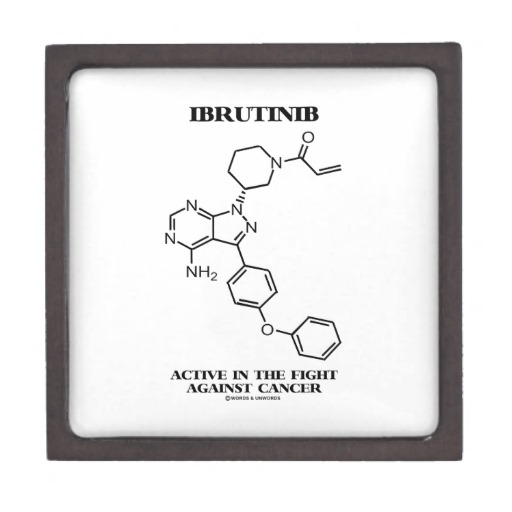
Even though the main objective of tumor surgery is to remove tumors as much as possible without disturbing the adjacent normal tissues, the task is very challenging in the operating room as neoplastic tissue is hard to distinguish from the adjacent healthy tissue. Thus, the portion of tumor still remained in the body after surgery causes recurrence, treatment failure, and poor outcome.
Surgery is an important treatment modality for brain tumors. Therefore, distinguishing normal tissue from tumor is extremely important for brain tumor surgery owing to the risk of damaging functional brain structures. Removal of healthy tissue can cause neurologic problems, but leaving tumor tissue behind can allow the cancer to grow and spread again. This is a major problem with glioblastoma multiforme (GBM), the most common form of malignant brain tumor in adults. Glioblastoma tumors grow quickly and are difficult to treat. The tumors infiltrate normal brain tissue and can’t be easily singled out. Therefore, tools designed to safely maximize the removal of tumor tissue are warranted. So far, experimental attempts to tell the difference between tumors and normal tissue during surgery have had limited success until recently, a new study by Ji et al. (2013) reported successful separation of tumor-infiltrated brain tissue from surrounding healthy tissue in mice using stimulated Raman scattering (SRS) microscopy. Ji and colleagues provided evidence that SRS microscopy can be used to delineate tumor tissue in a human GBM xenograft mouse model, both ex vivo and in vivo, and in human brain tumor surgical specimens.

Raman spectroscopy is a technique to study the interactions (vibrational, rotational, and other low-frequency modes) between matter and radiation in a system. It is named after the Indian noble laureate Dr. C.V. Raman (1930) who described the effect of light impinges upon a molecule and its interactions with the electron cloud and the bonds of that molecule. Chemical bonds in molecules have their own sets of vibration frequencies, and produce unique patterns of scattered light called Raman spectra. These spectra can be used as fingerprints to identify and differentiate different molecules in a complex environment. Developed on the concept of Raman spectroscopy, SRS is now emerging as an imaging technique to image biological tissues based on the intrinsic vibrational spectroscopy of their molecular components such as lipids, proteins, and DNA. Being free from the drawbacks of the dye-based methods, this label-free imaging technique exhibits high chemical selectivity enabling its use in complex biological applications including brain imaging.
To this end, Ji and colleagues used SRS microscopy to the problem of distinguishing protein-rich glioblastomas from more lipid-rich surrounding tissue and showed that it can be used to detect glioma ex vivo in human GBM xenograft mice, with results that correlated with the interpretation of hematoxylin and eosin (H&E)–stained slides by a surgical pathologist. Most importantly, this study demonstrated that SRS microscopy can detect extensive tumor infiltration in regions that appear grossly normal under standard bright-field conditions. This study suggests that SRS holds promise for improving the accuracy and effectiveness of cancer surgery. However, several challenges remain to be overcome including making a handheld surgical device with motion correction to acquire images from within a surgical cavity.
Reference:
Ji M, Orringer DA, Freudiger CW, Ramkissoon S, Liu X, Lau D, et al. Rapid, label-free detection of brain tumors with stimulated Raman scattering microscopy. Sci Transl Med. 2013;5:201ra119.



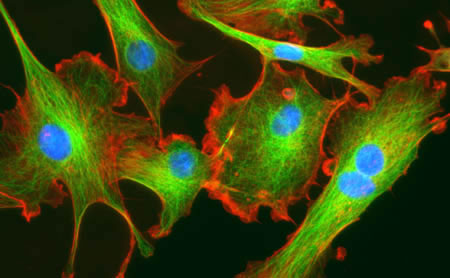
 recent edition of Nature Cell Biology Calvo et al. demonstrated that 6. Not only do the authors demonstrate that YAP/TAZ is active in CAFs, but YAP/TAZ is necessary for CAF development6. They show that CAF activation leads to matrix remodeling towards increased stiffness, via myosin light chain 2 (
recent edition of Nature Cell Biology Calvo et al. demonstrated that 6. Not only do the authors demonstrate that YAP/TAZ is active in CAFs, but YAP/TAZ is necessary for CAF development6. They show that CAF activation leads to matrix remodeling towards increased stiffness, via myosin light chain 2 (