Parkinson’s disease (PD) is a chronic neurodegenerative condition effecting dopaminergic neurons of the midbrain. PD manifests itself around age 50 with mainly motor symptoms, such as tremor (shaking), slowness of movement, rigidity and postural instability. Number of pharmaceutical agents (e.g., L-Dopa and MAO-B inhibitors) has been used for symptomatic relief in PD patients, but the ultimate therapy target is the replacement of degenerating dopaminergic neurons with new, healthy neurons.
 Cell replacement therapy for PD dates back to mid 80s with the transplantation of adrenal medullary tissue into patients’ striatum [1-3], which resulted only moderate improvements. At the same time, researchers in Sweden performed transplantation of fetal ventral mesencephalic tissue from aborted fetuses [4, 5]. These early studies observed important and persistent improvement based on numerous clinical outcomes. Moreover, postmortem examination of the brains of PD patients, who received ventral mesencephalic tissue transplantation, showed sustained survival of the graft and re-innervation of the striatum [6]. With the lift of federal funding ban on using fetal tissue for research and therapy by President Clinton in 1993, United States also began clinical trials utilizing fetal ventral mesencephalic tissue [7, 8]. Unfortunately, not only the patients didn’t display any significant improvements following transplantation in these trials, they developed additional abnormal, involuntary movements (i.e., graft-induced dyskinesia), due to surgery, which was also observed in other trials.
Cell replacement therapy for PD dates back to mid 80s with the transplantation of adrenal medullary tissue into patients’ striatum [1-3], which resulted only moderate improvements. At the same time, researchers in Sweden performed transplantation of fetal ventral mesencephalic tissue from aborted fetuses [4, 5]. These early studies observed important and persistent improvement based on numerous clinical outcomes. Moreover, postmortem examination of the brains of PD patients, who received ventral mesencephalic tissue transplantation, showed sustained survival of the graft and re-innervation of the striatum [6]. With the lift of federal funding ban on using fetal tissue for research and therapy by President Clinton in 1993, United States also began clinical trials utilizing fetal ventral mesencephalic tissue [7, 8]. Unfortunately, not only the patients didn’t display any significant improvements following transplantation in these trials, they developed additional abnormal, involuntary movements (i.e., graft-induced dyskinesia), due to surgery, which was also observed in other trials.
Close examination of the transplantation studies using fetal ventral mesencephalic tissue revealed few noteworthy outcomes:
1. Younger patients with newly developed pathology showed significant improvements over older patients with severe PD pathology.
2. Some patients showed continued improvements 3-4 years after surgery, while they did not display any benefits during the first year, indicating that the improvement in clinical parameters may take a while to appear over time. Regardless, it is clear that patients respond differently to the transplants of dopaminergic neurons, making the clinical outcomes fluctuate considerably.
3. Preparation of the fetal tissues, as well as selection of patients for transplantation, varied significantly from center to center carrying out the clinical trials, further indicating the need for standardizing tissue preparation, patient selection and implantation site.
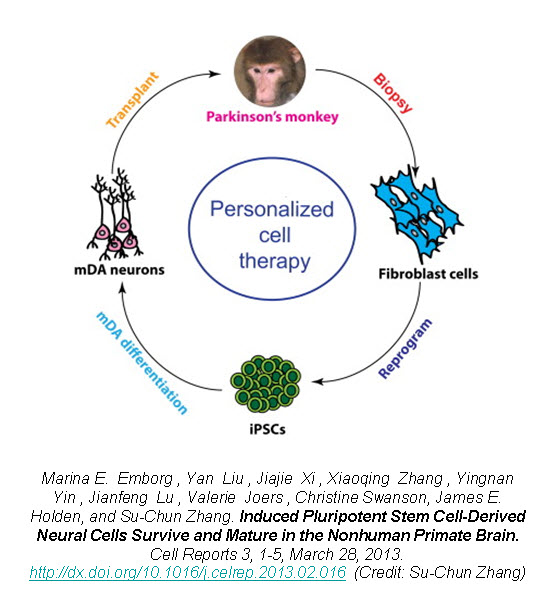
Compared to the aforementioned points, the use of fetal ventral mesencephalic tissue for grafting constitutes one of the biggest problems in cell based therapy for PD. It has been challenging to standardize the number and the quality of the fetal dopaminergic cells in graft preparations. Furthermore, the purity of the preparations also varies from batch to batch. Lastly, many ethical -and sometimes legal- issues surround fetal tissues/cells significantly limiting their clinical applicability. Do we have an alternative source that is free of these concerns/problems? The answer is yes, but not at the moment. With the isolation of human embryonic stem cells (hESCs) in 1998 and the introduction of human induced pluripotent stem cells (iPSCs) in 2007, stem cell derived dopaminergic neurons are at the top of everyone’s list when it comes to replacing degenerating neurons in PD. hESCs have been the primary source to produce dopaminergic neurons so far [9-11], but with the popularity and the advantages of iPSCs, the focus is more likely to shift to iPSC-derived dopaminergic neurons in future transplantation efforts.
Number of studies utilizing stem cell derived dopaminergic neurons in animal models of PD reported promising results over the years. However, we are far from using these cells in clinical trials. Many issues, such as long-term stability of the transplanted cells, sustained functional recovery, ability to re-innervate the host striatum, generation of GMP grade cells and long-terms safety especially with regards to tumor formation, remain to be determined. To be able to answer these concerns are critical for successful clinical translation of stem cell derived dopaminergic neurons. Nevertheless, the target is in front of everyone, and the field of regenerative medicine is moving at an incredible speed to reach it. It should also be noted that an increasing number of novel therapeutic approaches (e.g., gene therapy and growth factor infusions) have been under development -in addition to cell transplantations- with the aim of restoring dopaminergic function in PD patients.
While we are looking ahead with the promise of stem cell derived dopaminergic neurons for future of cell-based therapy in PD, there are many lessons to be learnt from the early clinical trials using fetal ventral mesencephalic tissue. There is no question that fetal dopamine neurons will serve as a reference and a standard against stem cell derived neurons for future clinical trials, since we know that the transplants survived, re-innervated the striatum, and generated adequate symptomatic relief in some patients for more than a decade following surgery. For PD patients, who are interested in cell-based therapy now, the decision of whether to wait for clinical trials utilizing stem cell derived neurons or to proceed with currently available fetal tissue grafts remains a somewhat difficult question and should take into consideration the aforementioned strengths and weaknesses of each approach.
References:
[1] Backlund EO, Granberg PO, Hamberger B, et al. Transplantation of adrenal medullary tissue to striatum in parkinsonism. First clini- cal trials. J Neurosurg 1985;62:169–173.
[2] Herrera-Marschitz M, Stromberg I, Olsson D, Ungerstedt U, Olson L. Adrenal medullary implants in the dopamine-denervated rat striatum. II. Acute behavior as a function of graft amount and location and its modulation by neuroleptics. Brain Res 1984;297:53–61.
[3] Madrazo I, Drucker-Colin R, Diaz V, Martinez-Mata J, Torres C, Becerril JJ. Open microsurgical autograft of adrenal medulla to the right caudate nucleus in two patients with intractable Parkinson’s disease. N Engl J Med 1987;316:831–834.
[4] Lindvall O, Brundin P, Widner H, et al. Grafts of fetal dopamine neurons survive and improve motor function in Parkinson’s dis- ease. Science 1990;247:574–577.
[5] Widner H, Tetrud J, Rehncrona S, et al. Bilateral fetal mesence- phalic grafting in two patients with parkinsonism induced by 1- methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). N Engl J Med 1992;327:1556–1563.
[6] Kordower JH, Rosenstein JM, Collier TJ, et al. Functional fetal nigral grafts in a patient with Parkinson’s disease: chemoanatomic, ultrastructural, and metabolic studies. J Comp Neurol 1996;370:203–230.
[7] Freed CR, Greene PE, Breeze RE, et al. Transplantation of embry- onic dopamine neurons for severe Parkinson’s disease. N Engl J Med 2001;344:710–719.
[8] Olanow CW, Goetz CG, Kordower JH, et al. A double-blind con- trolled trial of bilateral fetal nigral transplantation in Parkinson’s disease. Ann Neurol 2003;54:403–414.
[9] Lee SH, Lumelsky N, Studer L, Auerbach JM, McKay RD. Effi- cient generation of midbrain and hindbrain neurons from mouse embryonic stem cells. Nat Biotechnol 2000;18:675–679.
[10] Cho MS, Lee YE, Kim JY, et al. Highly efficient and large-scale generation of functional dopamine neurons from human embryonic stem cells. Proc Natl Acad Sci U S A 2008;105:3392–3397.
[11] Kawasaki H, Suemori H, Mizuseki K, et al. Generation of dopami- nergic neurons and pigmented epithelia from primate ES cells by stromal cell-derived inducing activity. Proc Natl Acad Sci U S A 2002;99:1580–1585.



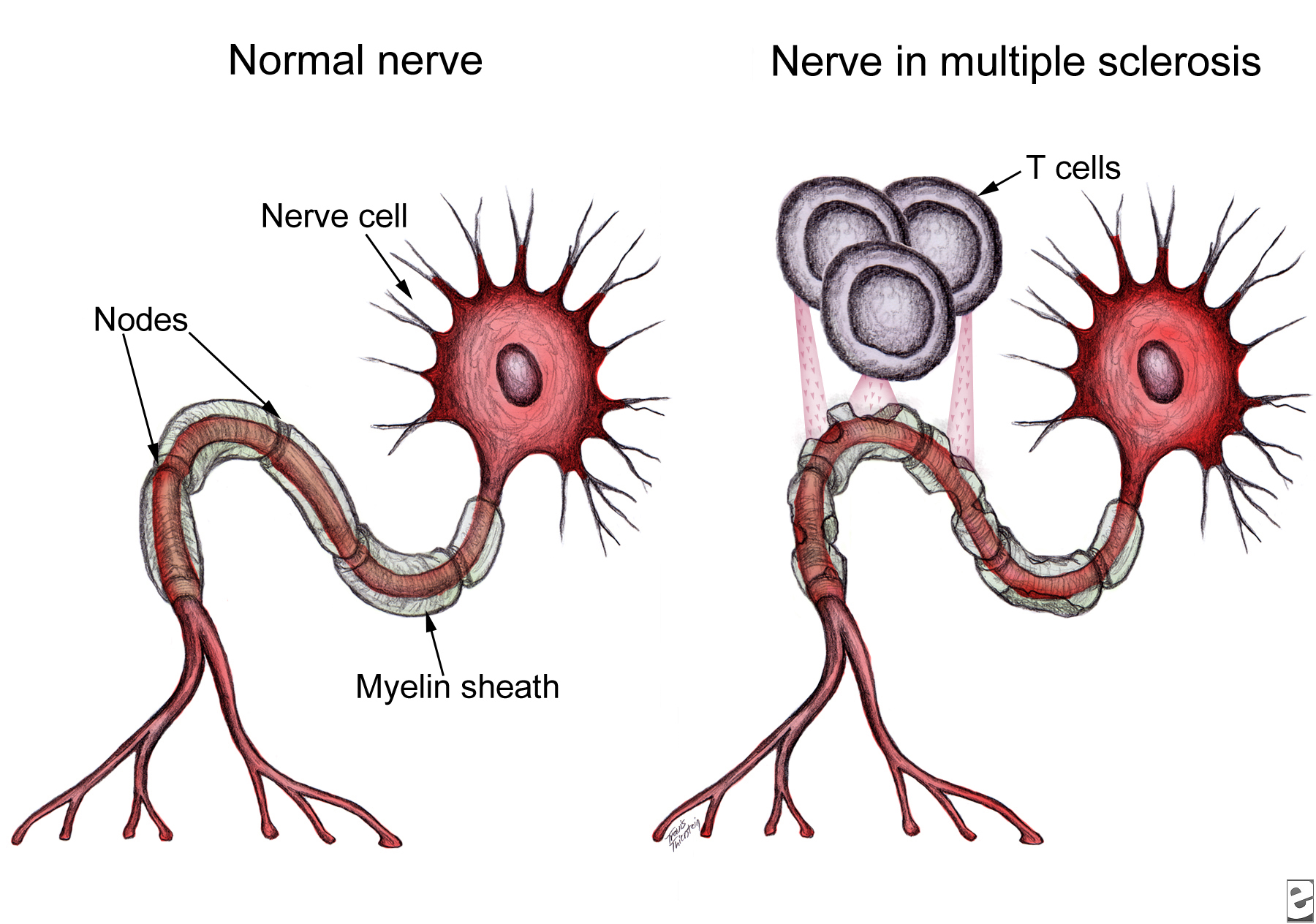

 These results support the epitope-spreading hypothesis, which indicates that MS patients make antibodies against one or a few myelin proteins, but as the disease progresses, the autoimmune response spreads to other myelin sheath epitopes. In their recent publication, Lutterotti et al. provide sufficient evidence necessitating emphasis not only on the specific target antigens, but also on the facility to inhibit epitope spreading, preferably prior to diversification of the
These results support the epitope-spreading hypothesis, which indicates that MS patients make antibodies against one or a few myelin proteins, but as the disease progresses, the autoimmune response spreads to other myelin sheath epitopes. In their recent publication, Lutterotti et al. provide sufficient evidence necessitating emphasis not only on the specific target antigens, but also on the facility to inhibit epitope spreading, preferably prior to diversification of the 
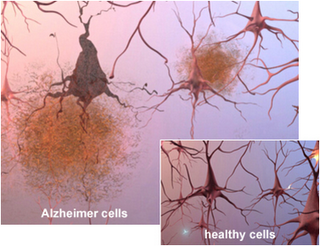
 With the increasing prevalence of neurodegenerative disorders in the aging population, it has become more and more important to understand the molecular pathways that regulate and advance these disorders. Due to the high level of complexity of the mammalian brain, it is very difficult to devise improved targeted treatments. The biggest limitation in neurodegenerative disease research being the lack of viable biomarkers for the elder population. Neurodegenerative disorders such as Alzheimer’s, Parkinson’s and polyglutamine diseases, share many pathogenic abnormalities such as the accumulation of misfolded proteins due to mutations rendering them resistant to degradation or over-expression of the wild type form.
With the increasing prevalence of neurodegenerative disorders in the aging population, it has become more and more important to understand the molecular pathways that regulate and advance these disorders. Due to the high level of complexity of the mammalian brain, it is very difficult to devise improved targeted treatments. The biggest limitation in neurodegenerative disease research being the lack of viable biomarkers for the elder population. Neurodegenerative disorders such as Alzheimer’s, Parkinson’s and polyglutamine diseases, share many pathogenic abnormalities such as the accumulation of misfolded proteins due to mutations rendering them resistant to degradation or over-expression of the wild type form.