
 The role of CD4+ FoxP3+ regulatory T cells (TREGS) in colorectal cancer (CRC) has continued to be unclear. TREGS act to suppress inflammatory mechanisms that are associated with tumor progression and can thus act to suppress the development of cancer. However, TREGS also function to inhibit anti-tumor T cell responses, thereby promoting cancer escape from immune surveillance. Many studies have been published on the frequencies of TREGS in the peripheral blood and tumors of CRC patients, but there is yet to be a consensus regarding the relationship between TREGS and disease outcome. In a report by Blatner et. al, the expression of RORγt in a subset of CD4+ FoxP3+ T cells was found to specifically mediate pathogenic pro-tumor activity compared with RORγt–CD4+FoxP3+ TREGS in CRC patients.
The role of CD4+ FoxP3+ regulatory T cells (TREGS) in colorectal cancer (CRC) has continued to be unclear. TREGS act to suppress inflammatory mechanisms that are associated with tumor progression and can thus act to suppress the development of cancer. However, TREGS also function to inhibit anti-tumor T cell responses, thereby promoting cancer escape from immune surveillance. Many studies have been published on the frequencies of TREGS in the peripheral blood and tumors of CRC patients, but there is yet to be a consensus regarding the relationship between TREGS and disease outcome. In a report by Blatner et. al, the expression of RORγt in a subset of CD4+ FoxP3+ T cells was found to specifically mediate pathogenic pro-tumor activity compared with RORγt–CD4+FoxP3+ TREGS in CRC patients.
CD4+ FoxP3+ cells have been classified into three functional populations based on the expression of CD45RA and FoxP3: CD45RA+FoxP3int, CD45RA–FoxP3int, and CD45RA–FoxP3high. The CD45RA–FoxP3high population exhibits the most suppressive activity of these subsets. In the study by Blatner et. al, the CD45RA–FoxP3high population was found to be specifically expanded in peripheral blood mononuclear cells (PBMCs) and within the tumor of CRC patients and increased with cancer stage. Because IL-17 expressing CD4+ FoxP3+ cells have been described in the gut and enhanced in patients with CRC and Crohn’s disease, the authors examined CD4+ FoxP3+ populations for expression of the TH17 transcription factor, RORγt.
In CRC patients, a large fraction of all three subsets of TREGS in PBMCs and in the tumor were found to express RORγt. Interestingly, when TREG populations were sorted from healthy donors versus CRC patients, CRC patient TREGS retained suppressive activity over T cell proliferation but had lost their ability to suppress mast cell degranulation. Expression of IL-17 was also found in a large percentage of CRC TREGS, in a fashion mutually exclusive from IL-10 expression.
To further explore the role of RORγt in CRC, APC∆468 polyposis-prone mice were crossed with mice deficient in RORγt. RORγt-/-APC∆468 mice were highly resistant to polyp development, had reduced expansion of splenic proinflammatory macrophages, myeloid-derived suppressor cells (MDSCs) and polyp-associated mast cells, compared with RORγt+APC∆468 mice. Interestingly, the effect of RORγt deficiency in APC∆468 mice was not phenocopied by the loss of IL-17. Instead, although IL-17 deficiency reduced the frequency of polyps, mast cell recruitment to polyps was enhanced, and ultimately IL-17-/-APC∆468 mice developed invasive lesions.
Overall, this study revealed several fascinating points: CD4+FoxP3+RORγt+ cells appear to be a pathogenic TREG subset that have lost their anti-inflammatory properties and are specifically expanded in CRC patients where they assist in disease progression. The function of RORγt was not synonymous with IL-17 in TREGS, indicating that other effects of RORγt contribute to the role of these cells in tumor pathogenesis. Thus, the roles and relationships between FoxP3, RORγt, and IL-17 in TREGS deserve further attention in CRC pathogenesis. Hopefully, a clearer understanding of this newly identified subset of RORγt+ TREGS and their role in CRC progression will enable much improved methodology for targeting specific TREGS populations in CRC and other disease settings.
Further Reading:
Expression of RORγt marks a pathogenic regulatory T cell subset in human colon cancer. Blatner NR, Mulcahy MF, Dennis KL, Scholtens D, Bentrem DJ, Phillips JD, Ham S, Sandall BP, Khan MW, Mahvi DM, Halverson AL, Stryker SJ, Boller AM, Singal A, Sneed RK, Sarraj B, Ansari MJ, Oft M, Iwakura Y, Zhou L, Bonertz A, Beckhove P, Gounari F, Khazaie K. Sci Transl Med. 2012 Dec 12;4(164):164ra159. doi: 10.1126/scitranslmed.3004566.
Translational mini-review series on Th17 cells: induction of interleukin-17 production by regulatory T cells. Afzali B, Mitchell P, Lechler RI, John S, Lombardi G. Clin Exp Immunol. 2010 Feb;159(2):120-30. doi: 10.1111/j.1365-2249.2009.04038.x. Epub 2009 Nov 11.
Inflammation-driven reprogramming of CD4+ Foxp3+ regulatory T cells into pathogenic Th1/Th17 T effectors is abrogated by mTOR inhibition in vivo. Yurchenko E, Shio MT, Huang TC, Da Silva Martins M, Szyf M, Levings MK, Olivier M, Piccirillo CA. PLoS One. 2012;7(4):e35572. doi: 10.1371/journal.pone.0035572. Epub 2012 Apr 24.
In colorectal cancer mast cells contribute to systemic regulatory T-cell dysfunction. Blatner NR, Bonertz A, Beckhove P, Cheon EC, Krantz SB, Strouch M, Weitz J, Koch M, Halverson AL, Bentrem DJ, Khazaie K. Proc Natl Acad Sci U S A. 2010 Apr 6;107(14):6430-5. doi: 10.1073/pnas.0913683107. Epub 2010 Mar 22.





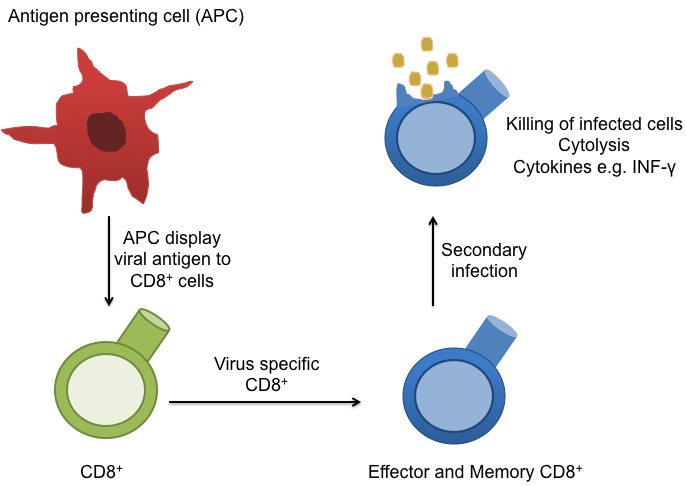
 circulate in the blood, lymph, and secondary lymphatic organs as they patrol for the presence of secondary infections. Recently, a class of effector memory CD8+ T cells has also been shown to migrate to and reside long-term in non-lymphoid tissues. These tissue resident memory T cells can quickly respond to tissue infections with their cognate pathogen. CD8+ T cells mainly function in killing of infected cells though cytolysis and by producing effector cytokines, including IFN-gamma to activate other immune cells. In a recent article in Nature Immunology,
circulate in the blood, lymph, and secondary lymphatic organs as they patrol for the presence of secondary infections. Recently, a class of effector memory CD8+ T cells has also been shown to migrate to and reside long-term in non-lymphoid tissues. These tissue resident memory T cells can quickly respond to tissue infections with their cognate pathogen. CD8+ T cells mainly function in killing of infected cells though cytolysis and by producing effector cytokines, including IFN-gamma to activate other immune cells. In a recent article in Nature Immunology, 
 cells in that they produce IL-5 and IL-13. RORγt+ ILCs aka ILC3s, include subsets that resemble TH17 and TH22 cells by producing IL-17 and IL-22, respectively, as well as a subset that produces both cytokines. Several recent articles have identified another class of ILCs in both humans and mice. These cells resemble TH1 cells in that they express T-bet/TBX21 and produce IFN-gamma, and are distinct from conventional NK cells found among peripheral blood mononuclear cells (PBMC). These newly characterized cellular subsets have been denoted as Type 1 ILCs (ILC1).
cells in that they produce IL-5 and IL-13. RORγt+ ILCs aka ILC3s, include subsets that resemble TH17 and TH22 cells by producing IL-17 and IL-22, respectively, as well as a subset that produces both cytokines. Several recent articles have identified another class of ILCs in both humans and mice. These cells resemble TH1 cells in that they express T-bet/TBX21 and produce IFN-gamma, and are distinct from conventional NK cells found among peripheral blood mononuclear cells (PBMC). These newly characterized cellular subsets have been denoted as Type 1 ILCs (ILC1).
 Activation of 41BB on T cells leads to enhanced T cell survival. Anti-41BB-agonistic antibodies have demonstrated significant anti-tumor activity in mice by enhancing anti-tumor cytotoxic T cell responses. Thus, there are currently several clinical trials underway exploring the efficacy of anti-41BB-agonist antibodies in several types of cancers, including melanoma, renal carcinoma, ovarian cancer, and lymphoma. In a previous study by the same group (
Activation of 41BB on T cells leads to enhanced T cell survival. Anti-41BB-agonistic antibodies have demonstrated significant anti-tumor activity in mice by enhancing anti-tumor cytotoxic T cell responses. Thus, there are currently several clinical trials underway exploring the efficacy of anti-41BB-agonist antibodies in several types of cancers, including melanoma, renal carcinoma, ovarian cancer, and lymphoma. In a previous study by the same group (
 -presentation is a DC-specialized mechanism by which antigens are taken up through endocytic and phagocytic pathways but presented in the context of MHC-class I, to activate antigen-specific cytotoxic CD8 T cells. Recent studies have sought to characterize the differences between the many tissue-associated DC subsets including their ability to cross-present antigen.
-presentation is a DC-specialized mechanism by which antigens are taken up through endocytic and phagocytic pathways but presented in the context of MHC-class I, to activate antigen-specific cytotoxic CD8 T cells. Recent studies have sought to characterize the differences between the many tissue-associated DC subsets including their ability to cross-present antigen.