
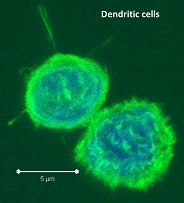
Dendritic cells (DC) are major antigen-presenting cells consisting of numerous heterogeneous subtypes. In humans, several subtypes of DCs have been identified in different tissues including peripheral blood, secondary lymphoid organs, and in the skin. In peripheral blood mononuclear cells (PBMC) and secondary lymphoid organs, these subtypes include BDCA1+, BDCA3+, and plasmacytoid DCs which differ functionally and in expression of various markers. So how do these many human DC subsets differ functionally?
Antigen cross -presentation is a DC-specialized mechanism by which antigens are taken up through endocytic and phagocytic pathways but presented in the context of MHC-class I, to activate antigen-specific cytotoxic CD8 T cells. Recent studies have sought to characterize the differences between the many tissue-associated DC subsets including their ability to cross-present antigen.
-presentation is a DC-specialized mechanism by which antigens are taken up through endocytic and phagocytic pathways but presented in the context of MHC-class I, to activate antigen-specific cytotoxic CD8 T cells. Recent studies have sought to characterize the differences between the many tissue-associated DC subsets including their ability to cross-present antigen.
In PBMC, DCs are quite rare, comprising only 1 – 2% of PBMCs. In previous blog posts, the generation of dendritic cells from PBMC monocytes and maturing and assaying monocyte-derived dendritic cells have been discussed. However, blood DCs and in vitro generated DCs may not represent the true physiological state of DCs present in secondary lymphoid organs where natural antigen cross-presentation and T cell activation occur in vivo.
In a recent study in The Journal of Experimental Medicine, Segura et. al explored the antigen cross-presentation versus phagocytic capabilities of human BDCA1+, BDCA3+, and plasmacytoid DC subsets compared with CD11c+HLADR+CD14+ macrophages that were all freshly isolated from healthy donor tonsils. The cross-presentation capabilities of different types of antigens, including necrotic dead cell antigens and soluble antigens were assessed.
For necrotic dead cell antigens, such as dead tumor cells, BDCA1+ and BDCA3+ DC subsets both took up antigens to a similar extent and were the most efficient in activating CD8+ T cell responses which were measured by IFN-gamma production in allogeneic mixed leukocyte reaction assays. Macrophages far exceeded the ability of any DC subsets in dead cell phagocytosis, but were extremely poor at cross-presentation and CD8+ T cell activation. Plasmacytoid DCs were the poorest at phagocytosis of dead cells, and were also unable to cross-present these antigens. For soluble antigens however, all three DC subsets (BDCA1+, BDCA3+, and plasmacytoid DC) efficiently cross-presented both shorter and longer soluble peptides, while macrophages continued to be poor at cross-presentation.
In an additional set of assays, the authors explored mechanisms that may contribute to the differential phagocytosis versus antigen cross-presentation of DC subsets and macrophages. Compared with macrophages which did not cross-present antigens, the endocytic compartments of cross-presenting DCs kept an alkaline pH and contained reactive oxygen species, and these DCs further were able to export internalized antigens to the cytosol where they can be loaded onto MHC-class I. Thus, while macrophages are efficient phagocytes, they are unable to process antigens to allow for cross-presentation.
In conclusion, understanding the capabilities of immune cells in different tissues is critical to discovering the full spectrum of cellular functions. DCs are a major target for vaccinations and immunotherapeutic strategies, and describing and understanding these subsets in vivo will lead to maximized success in immune modulating modalities.
Further Reading:
Similar antigen cross-presentation capacity and phagocytic functions in all freshly isolated human lymphoid organ-resident dendritic cells. Segura E, Durand M, Amigorena S. J Exp Med. 2013 Apr 8.
Cross-presentation by dendritic cells. Joffre OP, Segura E, Savina A, Amigorena S. Nat Rev Immunol. 2012 Jul 13;12(8):557-69. doi: 10.1038/nri3254. Review.
BDCA-2, BDCA-3, and BDCA-4: three markers for distinct subsets of dendritic cells in human peripheral blood. Dzionek, A., A. Fuchs, P. Schmidt, S. Cremer, M. Zysk, S. Miltenyi, D.W. Buck, and J. Schmitz. 2000. J. Immunol. 165:6037–6046.
Characterization of resident and migratory dendritic cells in human lymph nodes. Segura, E., J. Valladeau-Guilemond, M.H. Donnadieu, X. Sastre-Garau, V. Soumelis, and S. Amigorena. 2012. J. Exp. Med. 209:653– 660.
Gene family clustering identifies functionally associated subsets of human in vivo blood and tonsillar dendritic cells. Lindstedt, M., K. Lundberg, and C.A. Borrebaeck. 2005. J. Immunol. 175:4839–4846.
Functional specializations of human epidermal Langerhans cells and CD14+ dermal dendritic cells. Klechevsky, E., R. Morita, M. Liu, Y. Cao, S. Coquery, L. Thompson- Snipes, F. Briere, D. Chaussabel, G. Zurawski, A.K. Palucka, et al. 2008. Immunity. 29:497–510.
Characterization of dermal dendritic cells obtained from normal human skin reveals phenotypic and functionally distinctive subsets. Nestle, F.O., X.G. Zheng, C.B. Thompson, L.A. Turka, and B.J. Nickoloff. 1993. J. Immunol. 151:6535–6545.



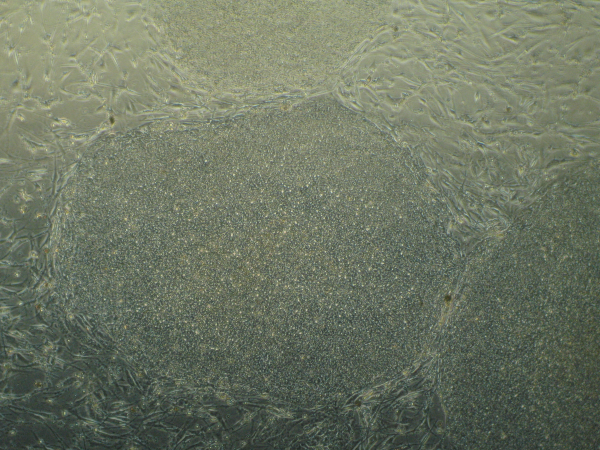
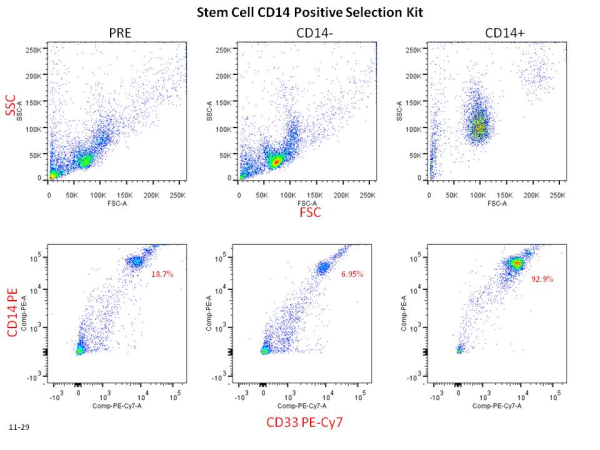
 Recently, in Stem Cells, Yamanaka’s group reported a protocol that increased the efficiency of iPSC induction from CD34+ cord blood and peripheral blood
Recently, in Stem Cells, Yamanaka’s group reported a protocol that increased the efficiency of iPSC induction from CD34+ cord blood and peripheral blood 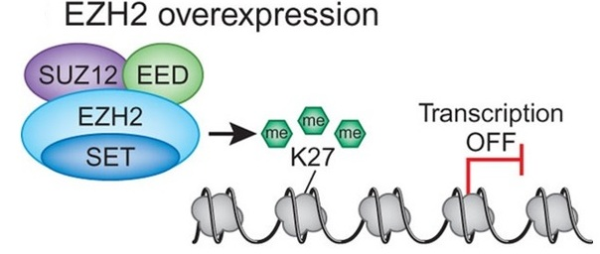
 d breast cancer. In both cases increased expression was found to be associated with tumor invasiveness, metastasis, and poor clinical outcome. In addition, elevated expression of EZH2 is also reported in several other tumors including gastric, lung, bladder, and endometrial cancer. Gain of functions as a result of acquired mutations in EZH2 was reported in lymphoma and meyloid neoplasms. The best characterized mechanism by which EZH2 exerts its oncogenic function is by transcriptional repression of genes via its histone methyltransferase activity. Genes which get transcriptionally repressed by EZH2 include tumor suppressor genes ARF, p57KIP2, FBXO32, p27, and BRCA1. In addition, this enzyme also activates transcription of gene CCND1 (cyclin D1) driving cell-cycle progression.
d breast cancer. In both cases increased expression was found to be associated with tumor invasiveness, metastasis, and poor clinical outcome. In addition, elevated expression of EZH2 is also reported in several other tumors including gastric, lung, bladder, and endometrial cancer. Gain of functions as a result of acquired mutations in EZH2 was reported in lymphoma and meyloid neoplasms. The best characterized mechanism by which EZH2 exerts its oncogenic function is by transcriptional repression of genes via its histone methyltransferase activity. Genes which get transcriptionally repressed by EZH2 include tumor suppressor genes ARF, p57KIP2, FBXO32, p27, and BRCA1. In addition, this enzyme also activates transcription of gene CCND1 (cyclin D1) driving cell-cycle progression.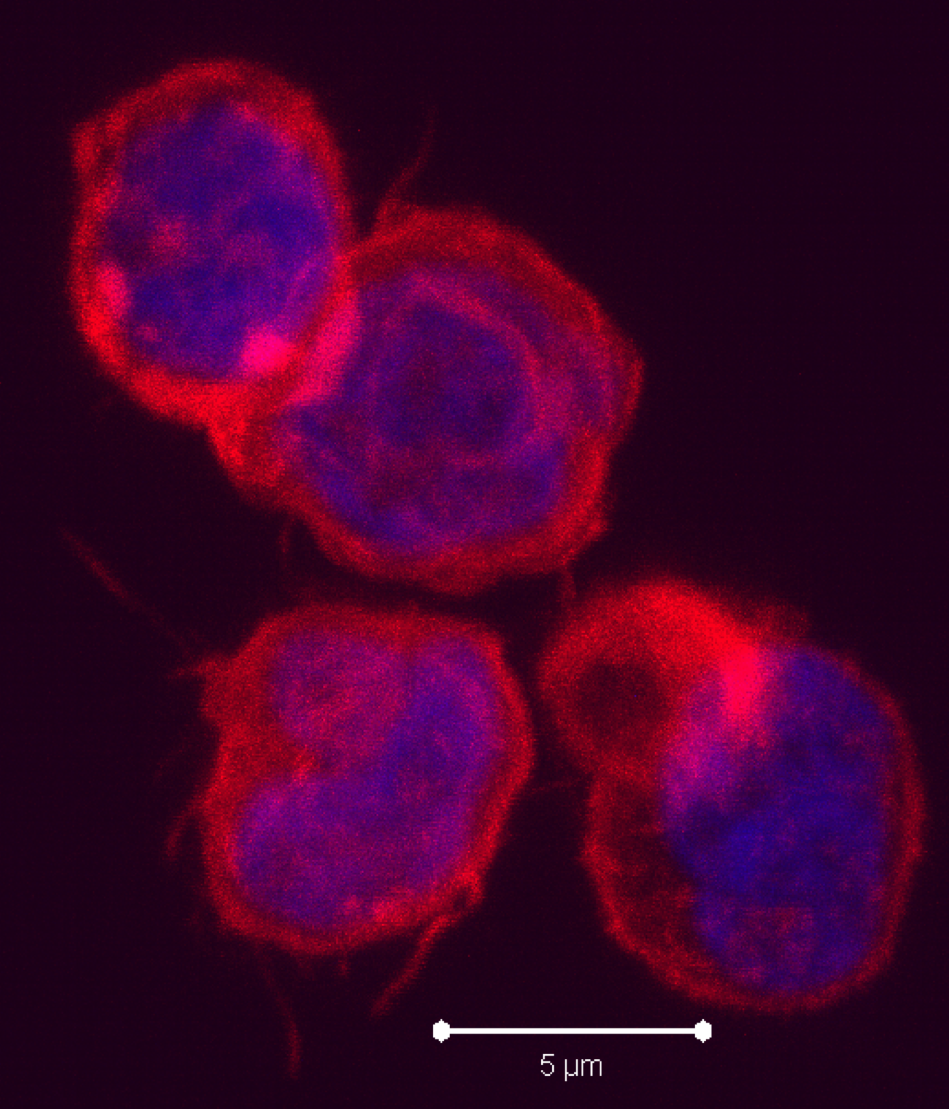
 9)
9)


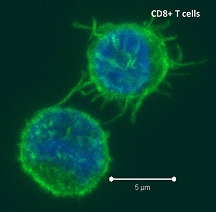
 Negative regulatory CD4 T cells are well characterized and highly studied. However their CD8 counterparts are not well defined, particularly in humans. Regulatory CD8 T cells suppress activated CD4 T cells and have proposed roles in various human diseases including multiple sclerosis, ovarian carcinoma and infection with HIV, and many subsets have been described using various markers. In a recent issue of PLoS One, Hu et. al, describe a population of CD3+CD8+CD161−CD56+ T cells within human peripheral blood mononuclear cells (PBMC) that exhibit a cytolytic negative regulatory function.
Negative regulatory CD4 T cells are well characterized and highly studied. However their CD8 counterparts are not well defined, particularly in humans. Regulatory CD8 T cells suppress activated CD4 T cells and have proposed roles in various human diseases including multiple sclerosis, ovarian carcinoma and infection with HIV, and many subsets have been described using various markers. In a recent issue of PLoS One, Hu et. al, describe a population of CD3+CD8+CD161−CD56+ T cells within human peripheral blood mononuclear cells (PBMC) that exhibit a cytolytic negative regulatory function.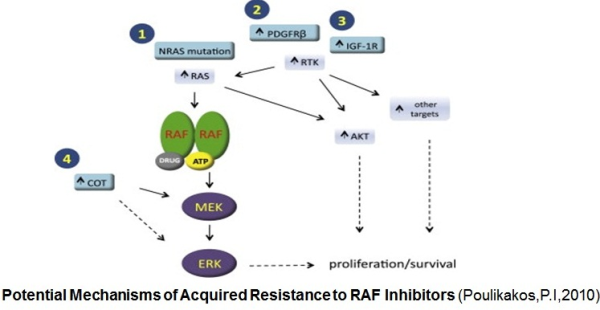
 ibitors. Activation of the MAPK signaling and increased expression of C-RAF (an isoform of B-RAF) was noted in melanoma cells resistant to B-RAF inhibitors. In this study Villanueva et al. (2010 ) also observed constitutive activation of the insulin-like growth factor receptor 1 (IFGR1), a receptor tyrosine kinase (RTK) in the resistant cells. As IGFR1 activates PI3K/Akt signaling, combined treatment of PI3K and MEK inhibitors resulted in resistance reversal. Increased levels of IGFR1 was also observed in melanoma patients failing vemurafenib suggesting activation of PI3K/Akt signaling via IGFR1 could limit the efficacy of B-RAF inhibitors in the clinic. Up-regulation of other RTKs was also found to be associated with the acquired resistance to vemurafenib. Tumor biopsies of melanoma patients failing vemurafenib exhibited over-expression of the platelet derived growth factor receptor-β (Nazarian et al., 2010).
ibitors. Activation of the MAPK signaling and increased expression of C-RAF (an isoform of B-RAF) was noted in melanoma cells resistant to B-RAF inhibitors. In this study Villanueva et al. (2010 ) also observed constitutive activation of the insulin-like growth factor receptor 1 (IFGR1), a receptor tyrosine kinase (RTK) in the resistant cells. As IGFR1 activates PI3K/Akt signaling, combined treatment of PI3K and MEK inhibitors resulted in resistance reversal. Increased levels of IGFR1 was also observed in melanoma patients failing vemurafenib suggesting activation of PI3K/Akt signaling via IGFR1 could limit the efficacy of B-RAF inhibitors in the clinic. Up-regulation of other RTKs was also found to be associated with the acquired resistance to vemurafenib. Tumor biopsies of melanoma patients failing vemurafenib exhibited over-expression of the platelet derived growth factor receptor-β (Nazarian et al., 2010).
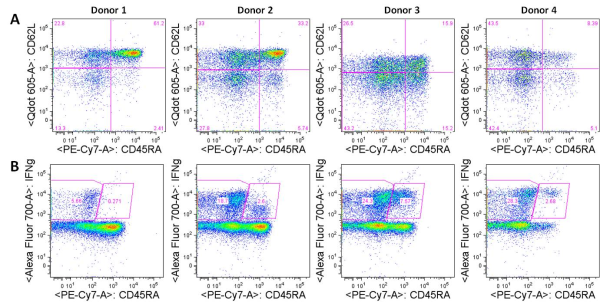
 Flow cytometry has been around since the 1950s when Wallace Coulter developed the first flow cytometry device and fluorescence-based flow cytometry was introduced in 1968 by Wolfgang Göhde. Since then, fluorescence-based flow cytometry and fluorescence-activated cell sorting (FACS) have blown up to become a mainstay of analytical scientific approaches in every field of cell biology, especially immunology. However, the dominance of fluorescence-based flow cytometry for analytical cellular biology may change with the recent introduction of a new technology: Time of Flight Mass Cytometry (CyTOF).
Flow cytometry has been around since the 1950s when Wallace Coulter developed the first flow cytometry device and fluorescence-based flow cytometry was introduced in 1968 by Wolfgang Göhde. Since then, fluorescence-based flow cytometry and fluorescence-activated cell sorting (FACS) have blown up to become a mainstay of analytical scientific approaches in every field of cell biology, especially immunology. However, the dominance of fluorescence-based flow cytometry for analytical cellular biology may change with the recent introduction of a new technology: Time of Flight Mass Cytometry (CyTOF).