Since the mid-1880’s, scientists and clinicians have studied the concept of sepsis. Investigations started with physician-scientists such as Ignaz Semmelweiss, Joseph Lister, Hugo Schottmüller and Louis Pasteur, initiating the fields of immunology and diseases related to immunology that continue even to today. Unfortunately, while we increase our knowledge about sepsis (also known as systemic inflammatory response syndrome; SIRS), diagnosis and treatment remain difficult1,2. This is seen as recently as last year, when an infection of a 12-year-old boy , Rory Staunton, lead to fatal septic shock after doctors failed to see the early signs of SIRS3. His death initiated the formation of the Rory Staunton Foundation and subsequent reforms of emergency protocols taken in his home state of New York (labeled as “Rory’s Laws”)3.
 However, hospital policy reforms are just part of the issue; sepsis affects more than 700,000 people in North America every year, with a 30-50% mortality rate4. To date, there are no FDA-approved drugs to fight SIRS4. In fact, after 20 years of intense research into translational medicine for sepsis, none of the proposed treatment approaches have had enough clinical efficacy to be used as treatment paradigms1. Action must be taken to not only change the methodologies of care for sepsis, but to better understand the biological mechanisms of sepsis. From this understanding, we may be able to have more accurate detection of sepsis as well as more effective therapeutics.
However, hospital policy reforms are just part of the issue; sepsis affects more than 700,000 people in North America every year, with a 30-50% mortality rate4. To date, there are no FDA-approved drugs to fight SIRS4. In fact, after 20 years of intense research into translational medicine for sepsis, none of the proposed treatment approaches have had enough clinical efficacy to be used as treatment paradigms1. Action must be taken to not only change the methodologies of care for sepsis, but to better understand the biological mechanisms of sepsis. From this understanding, we may be able to have more accurate detection of sepsis as well as more effective therapeutics.
Sepsis occurs when an infection causes a systemic inflammatory response5. This disease causes an imbalance of the immune system, leading to changes in the hemodynamics of the host, resulting in coagulation, heart ischemia, and multi-organ failure1. Originally thought to be caused by overactivation of the innate immune system, many patients did not die from the initial onslaught of inflammation, but from later stages of immune system suppression (also known as “immunoparalysis”) which allowed opportunistic viruses and bacteria to take over the host1. The roles of sepsis may be analyzed by observing the different cell types it affects: the innate system, the adaptive immune system, and non-immune cells.
Since sepsis is due to bacterial and viral infection, the innate immune system is the most well studied aspect of the pathogenesis of sepsis. During SIRS the innate immune cells are pathologically affected: macrophages, dendritic cells and natural killer (NK) cells. During the initial period of infection, increased amounts of pathogen-associated molecular pattern (PAMPs) and damage-associate molecular pattern (DAMP) molecules are found in the host organism. Toll-like receptors (TLRs), such as TLR4 and TLR9, in the innate immune cells recognize these molecules and confer a host inflammatory response. The inflammation leads to an increased presence of adhesion molecules on both innate and adaptive immune cells4 indicating the desire to enter the site of infection to confer an immune response. Along the same lines, activation of the complement cascade occurs, leading to increased amounts of C5a protein. The upregulated C5a protein levels cause increased migration of innate immune cells and increased phagocytic ability of macrophages4.
Unfortunately, prolonged exposure of macrophages to C5a leads to dysregulation of macrophage activation and, eventually, apoptosis1. During this apoptotic stage, macrophages release high-mobility group protein B1 (HMGB1) which may lead to increased inflammation5. On the other hand, in neutrophils, sepsis is known to cause an abnormally long proliferation period5 which could lead to organ damage and increased inflammation. Surprisingly, sustained activation of neutrophils may also contribute to the immunoparalysis by sepsis because activated neutrophils increase reactive oxygen species, known to cause immunosuppression, and macrophages that eat apoptotic neutrophils express anti-inflammatory cytokines5.  Prolonged sepsis also decreases the pro-inflammatory ability of dendritic cells; SIRS increases depletion of splenic and myeloid dendritic cells, where the remaining dendritic cells are functionally deficient4. NK cells, originally shown to play a role in anti-viral immune responses but are also proposed to play a role in bacterial infection responses, have two distinct roles in different phases of sepsis. In the early phases, NK cells are thought to contribute to the overactive immune responses and systemic inflammation4 however, in later phases of sepsis, NK cells may be compromised which could lead to secondary bacterial or viral infections, thus exacerbating the inflammatory response during SIRS4.
Prolonged sepsis also decreases the pro-inflammatory ability of dendritic cells; SIRS increases depletion of splenic and myeloid dendritic cells, where the remaining dendritic cells are functionally deficient4. NK cells, originally shown to play a role in anti-viral immune responses but are also proposed to play a role in bacterial infection responses, have two distinct roles in different phases of sepsis. In the early phases, NK cells are thought to contribute to the overactive immune responses and systemic inflammation4 however, in later phases of sepsis, NK cells may be compromised which could lead to secondary bacterial or viral infections, thus exacerbating the inflammatory response during SIRS4.
 In addition to dysregulation of the innate immune system, the cells that make up the adaptive immune system, T-cells and B-cells, are also affected by sepsis. Sepsis decreases overall T-cell receptor function, and moves Th1 (proinflammatory) T-cells toward a Th2 (immunosuppressive) response1. In addition, CD25+Foxp3+ T-cells, also known as regulatory T-cells, are increased during SIRS5. Interferon-gamma, a proinflammatory cytokine expressed during sepsis, causes an increase in innate response activator (IRA) B Cells, B-cells that recognize PAMPs, impair infection clearance, and accelerate septic shock4,6. During sepsis, the increase of C5a causes T-Cell and B-cell apoptosis4, which leads to immunosuppression4.
In addition to dysregulation of the innate immune system, the cells that make up the adaptive immune system, T-cells and B-cells, are also affected by sepsis. Sepsis decreases overall T-cell receptor function, and moves Th1 (proinflammatory) T-cells toward a Th2 (immunosuppressive) response1. In addition, CD25+Foxp3+ T-cells, also known as regulatory T-cells, are increased during SIRS5. Interferon-gamma, a proinflammatory cytokine expressed during sepsis, causes an increase in innate response activator (IRA) B Cells, B-cells that recognize PAMPs, impair infection clearance, and accelerate septic shock4,6. During sepsis, the increase of C5a causes T-Cell and B-cell apoptosis4, which leads to immunosuppression4.
Sepsis also has non-immunological effects on the patient. During sepsis, coagulation becomes more pronounced. This increase in coagulation and pre-coagulation states may lead to ischemic injury5. Sepsis also causes cytopathic hypoxia5 and cardiomyopathy4. Interestingly, SIRS also plays a role on the autonomic nervous system, with increased apoptosis of the adrenal medullary cells, there is a dysregulation of the endocrine system that regulates the autonomic nervous system1.
Currently, there are several studies looking into the pathogenesis of sepsis with the goal being effective treatment. In terms of pathogenesis, recent studies looking at the role of STIM17 and PI3K activation in the initiation of sepsis5. Host deficiencies leading to sepsis are also being studied. Deficiencies, including, zinc8 and ADAMS-T5 deficiencies, will hopefully elucidate more on the dysregulation found in sepsis. Currently, medical scientists are working with clinicians to study the more nuanced roles of hyper-responsiveness versus system immune suppression4. From these studies, multiple immunotherapies have been proposed: dendritic cell implantation4, regulatory T-cell implantation 4, and the modification of the host immune system by using TLR antagonists4, injections of the proinflammatory cytokines IL-15 and IL-174, and limiting adaptive immune system exhaustion by blocking PD-14. However, several of these studies, such as T-reg implantation, and using TLR antagonists have had limited clinical efficacy and have been prematurely terminated4. This may be due to the lack of an appropriate model to study human sepsis in the pre-clinical setting.
The limitations of current research methodologies for sepsis have shown the need for reform in SIRS research. Recently an article by Seok et al in the Proceedings of the National Academy of Sciences elucidated the limitations of using mouse models to studying human inflammation9. In this article, Seok et al list the differences in temporal responses, gene signatures, and regulated pathways involved inflammatory response following various insults9. The reasons for  differences in the inflammatory response may be described by the evolutionary differences between mouse and human immune systems, the inbred nature of the mice used, and the tendency to focus on a single mechanism in mouse models when there is great overlap of immune responses within a host system. These authors propose studying more of the genetic and epigenetic changes of patient samples during sepsis to find appropriate mouse models for study9. Furthermore, they propose in vitro recapitulation of the inflammation response in diseased tissue9. Unfortunately, the in vitro model is limited and this reductionist approach may miss a key cell, cellular event, or special/temporal arrangement that may be vital to pathogenesis of sepsis.
differences in the inflammatory response may be described by the evolutionary differences between mouse and human immune systems, the inbred nature of the mice used, and the tendency to focus on a single mechanism in mouse models when there is great overlap of immune responses within a host system. These authors propose studying more of the genetic and epigenetic changes of patient samples during sepsis to find appropriate mouse models for study9. Furthermore, they propose in vitro recapitulation of the inflammation response in diseased tissue9. Unfortunately, the in vitro model is limited and this reductionist approach may miss a key cell, cellular event, or special/temporal arrangement that may be vital to pathogenesis of sepsis.
The use of non-human primates as a sepsis model may be more accurate than either of these, but the ethical and cost-effective issues of using non-human primates remains a deterrent. A final model that may be of use to sepsis researchers in the future is the use of the humanized mouse model; a severely immunocompromised mouse host that has had a recapitulation of the adaptive and/or innate immune systems for in vivo study of human pathologies. The use of humanized mice for the study of sepsis was proposed by Unsinger et al, where they used two-day-old NOD-scid IL2 receptor-gamma knockout mice and transplanted them with hCD34+ enriched hematopoietic cord blood stem cells. After establishment of the human immune system, mice were treated with cecal ligation puncture (CLP) model of intra-abdominal peritonitis and assayed for immune response changes10. These mice developed a functional human innate and adaptive immune system with recapitulation of the human immune response to sepsis. However, while there still remain differences in the humanized mouse model versus the actual human immune response, such as differences in the bacterial flora that is found in the bowels10, this model nonetheless provides a useful tool which, along with more genetic and epigenetic information from patient studies, will lead to more impactful pre-clinical studies and possibly FDA-approved drugs to treat sepsis in the clinics.
References:
1. Rittirsch, D., Flierl, M. A. & Ward, P. A. Harmful molecular mechanisms in sepsis. Nat Rev Immunol 8, 776-787, doi:10.1038/nri2402 (2008).
2. Stearns-Kurosawa, D. J., Osuchowski, M. F., Valentine, C., Kurosawa, S. & Remick, D. G. The pathogenesis of sepsis. Annu Rev Pathol 6, 19-48, doi:10.1146/annurev-pathol-011110-130327 (2011).
3. Dwyer, J. Death of a Boy Prompts New Medical Efforts Nationwide, October 26, 2012).
4. Ward, P. A. & Bosmann, M. A historical perspective on sepsis. Am J Pathol 181, 2-7, doi:10.1016/j.ajpath.2012.05.003 (2012).
5. Cinel, I. & Opal, S. M. Molecular biology of inflammation and sepsis: a primer. Crit Care Med 37, 291-304, doi:10.1097/CCM.0b013e31819267fb (2009).
6. Rauch, P. J. et al. Innate response activator B cells protect against microbial sepsis. Science 335, 597-601, doi:10.1126/science.1215173
7. Gandhirajan, R. K. et al. Blockade of NOX2 and STIM1 signaling limits lipopolysaccharide-induced vascular inflammation. J Clin Invest, doi:10.1172/jci65647 (2013).
8. Liu, M. J. et al. ZIP8 Regulates Host Defense through Zinc-Mediated Inhibition of NF-κB. Cell Rep, doi:10.1016/j.celrep.2013.01.009 (2013).
9. Seok, J. et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci U S A, doi:10.1073/pnas.1222878110 (2013).
10. Unsinger, J., McDonough, J. S., Shultz, L. D., Ferguson, T. A. & Hotchkiss, R. S. Sepsis-induced human lymphocyte apoptosis and cytokine production in “humanized” mice. J Leukoc Biol 86, 219-227, doi:10.1189/jlb.1008615 (2009).




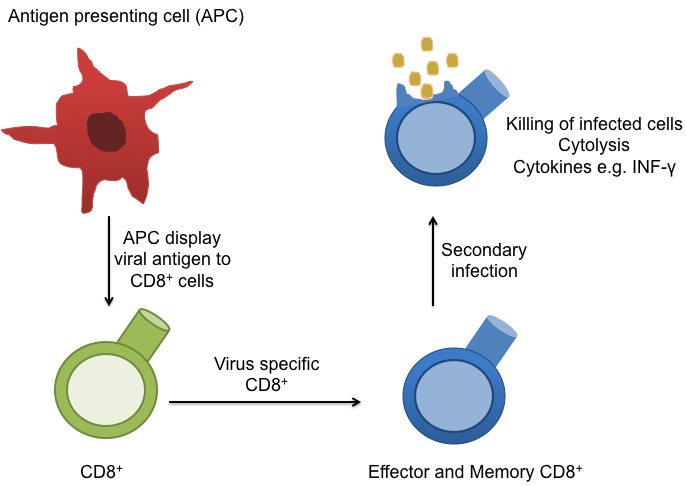
 circulate in the blood, lymph, and secondary lymphatic organs as they patrol for the presence of secondary infections. Recently, a class of effector memory CD8+ T cells has also been shown to migrate to and reside long-term in non-lymphoid tissues. These tissue resident memory T cells can quickly respond to tissue infections with their cognate pathogen. CD8+ T cells mainly function in killing of infected cells though cytolysis and by producing effector cytokines, including IFN-gamma to activate other immune cells. In a recent article in Nature Immunology,
circulate in the blood, lymph, and secondary lymphatic organs as they patrol for the presence of secondary infections. Recently, a class of effector memory CD8+ T cells has also been shown to migrate to and reside long-term in non-lymphoid tissues. These tissue resident memory T cells can quickly respond to tissue infections with their cognate pathogen. CD8+ T cells mainly function in killing of infected cells though cytolysis and by producing effector cytokines, including IFN-gamma to activate other immune cells. In a recent article in Nature Immunology, 

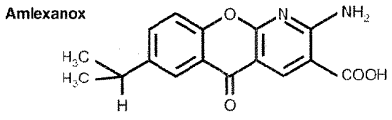
 Can amlexanox, a small molecule drug that has been approved for asthma, allergic rhinitis and aphthous ulcers be an effective treatment for obesity? Many are excited about this prospective presented in a recent article in Nature Medicine by Reilly et. al. as simply Googling “amlexanox” and “obesity” resulted in a plethora of news articles on this report. Interestingly, the two highly related proteins allegedly inhibited by amlexanox that led to this result in mice are the IRF3-kinases, TBK1 (TANK-binding Kinase-1, T2K, NAK) and IKK-ε (IκB kinase-epsilon, IKK-i), whose major known functions are during pathogen infections: the activation of IRF-3 (interferon regulatory kinase-3), the major transcription factor regulating expression of interferon-β (IFNβ).
Can amlexanox, a small molecule drug that has been approved for asthma, allergic rhinitis and aphthous ulcers be an effective treatment for obesity? Many are excited about this prospective presented in a recent article in Nature Medicine by Reilly et. al. as simply Googling “amlexanox” and “obesity” resulted in a plethora of news articles on this report. Interestingly, the two highly related proteins allegedly inhibited by amlexanox that led to this result in mice are the IRF3-kinases, TBK1 (TANK-binding Kinase-1, T2K, NAK) and IKK-ε (IκB kinase-epsilon, IKK-i), whose major known functions are during pathogen infections: the activation of IRF-3 (interferon regulatory kinase-3), the major transcription factor regulating expression of interferon-β (IFNβ).
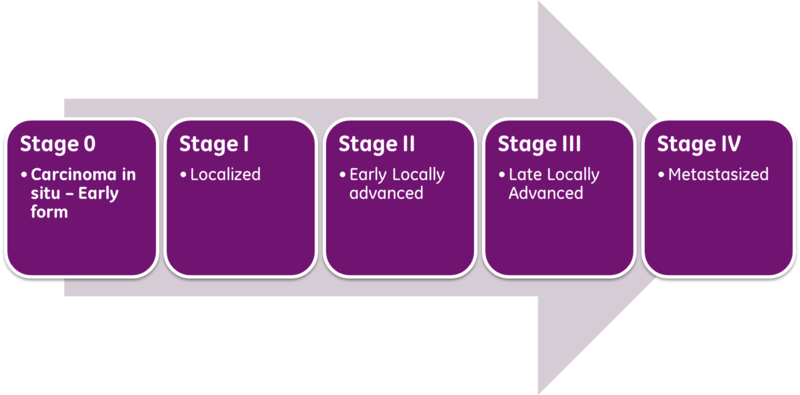
 Classical prognosis of cancer patients utilizes the AJCC/UICC (American Joint Committee on Cancer / International Union Against Cancer) “TNM” classification system, in which T (Tumor) is indicative of primary tumor size and invasion properties, N (Nodes) indicates the extent of tumor invasion into draining and regional lymph nodes, and M (Metastasis), describes the presence and extent of metastatic lesions at diagnosis. The combinations of these parameters are then used to assess a patient’s stage at diagnosis and predict patient outcome. The exact parameter definitions vary for each cancer type.
Classical prognosis of cancer patients utilizes the AJCC/UICC (American Joint Committee on Cancer / International Union Against Cancer) “TNM” classification system, in which T (Tumor) is indicative of primary tumor size and invasion properties, N (Nodes) indicates the extent of tumor invasion into draining and regional lymph nodes, and M (Metastasis), describes the presence and extent of metastatic lesions at diagnosis. The combinations of these parameters are then used to assess a patient’s stage at diagnosis and predict patient outcome. The exact parameter definitions vary for each cancer type.
 Interferons, including type I (IFNα/β) and type II (IFNγ) are known to be critical for mediating multiple aspects of tumor immunity, by targeting both immune cells for activation, and cancer cells for expression of MHC and genes associated with growth arrest and apoptosis. Thus, expression of cytokines such as IFNγ by immune cells has been shown to be critical in anti-tumor immune responses. IFNγ is one of the major effector cytokines of CD4+ TH1 cells and cytotoxic CD8+ T cells. However, the full effects of IFNγ as well as other TH1 cytokines in mediating anti-tumor effects have not been fully elucidated.
Interferons, including type I (IFNα/β) and type II (IFNγ) are known to be critical for mediating multiple aspects of tumor immunity, by targeting both immune cells for activation, and cancer cells for expression of MHC and genes associated with growth arrest and apoptosis. Thus, expression of cytokines such as IFNγ by immune cells has been shown to be critical in anti-tumor immune responses. IFNγ is one of the major effector cytokines of CD4+ TH1 cells and cytotoxic CD8+ T cells. However, the full effects of IFNγ as well as other TH1 cytokines in mediating anti-tumor effects have not been fully elucidated.
 In a study published in the February 2013 issue of Immunity, Su. et. al., characterized the CD4+ T cell repertoire from adult human peripheral blood mononuclear cells (PBMC) and made a fascinating observation: that
In a study published in the February 2013 issue of Immunity, Su. et. al., characterized the CD4+ T cell repertoire from adult human peripheral blood mononuclear cells (PBMC) and made a fascinating observation: that 
