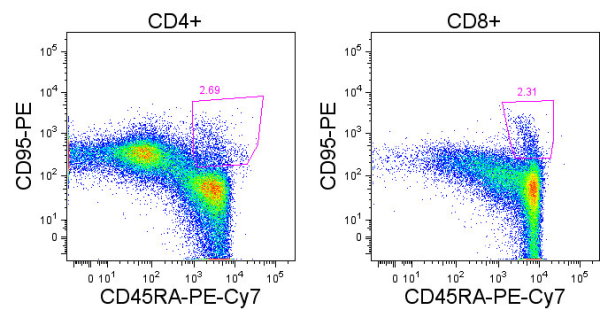
 The anti-tumor effectiveness of IFNγ-producing CD4+ TH1 cells and CD8+ TC1 cells is well accepted. However, the role of IL-17 producing CD4+ (TH17) and CD8+ T cells (TC17) in promoting or inhibiting tumor growth remains unclear, as various studies have shown both tumor-inhibiting and tumor-promoting functions of these cell types. Thus, context is a key determinant factor in the role of IL-17 producing T cell subsets in tumor immune responses.
The anti-tumor effectiveness of IFNγ-producing CD4+ TH1 cells and CD8+ TC1 cells is well accepted. However, the role of IL-17 producing CD4+ (TH17) and CD8+ T cells (TC17) in promoting or inhibiting tumor growth remains unclear, as various studies have shown both tumor-inhibiting and tumor-promoting functions of these cell types. Thus, context is a key determinant factor in the role of IL-17 producing T cell subsets in tumor immune responses.
In the current February 15, 2013 issue of The Journal of Immunology, Yu. et. al. compared the role of adoptively transferred tumor-specific TC1 and TC17 cells in controlling tumor growth. In this murine model, anti-gp100 T cells from Pmel-1 TCR transgenic mice were polarized ex-vivo into TC1 or TC17 phenotypes, and adoptively transferred into luciferase-expressing B16F10 melanoma lung metastatic tumor-bearing mice that had undergone total body irradiation.
The efficacy of anti-tumor TC1 or TC17 cells in mediating tumor regression was monitored by measuring tumor burden by luciferase luminescence and overall survival. By these measures, both TC1 and TC17 cells exhibited anti-tumor activity, however TC1 cells were superior. TC1 cells completely inhibited tumor growth, while TC17 cells delayed tumor growth but were ultimately unable to control the tumor.
Adoptively transferred TC1 cells were found to produce only IFNγ but not IL-17, while TC17 cells expressed high levels of IL-17 and could also differentiate into IFNγ-producing cells, as is known for these cell subsets. In a fascinating observation, the authors found that eliminating IFNγ-responsiveness in tumor cells completely reversed the relative efficacy of TC1 and TC17 cells. IFNγ-responsiveness in tumor cells was required for TC1 mediated anti-tumor activity, indicating a critical role for IFNγ-responsive genes in promoting tumor cell recognition by TC1 cells and/or growth inhibition or sensitivity to apoptosis. However, when tumor cells could no longer respond to IFNγ, TC17 cells were now able to induce complete tumor regression. In cytokine-neutralization assays, in vivo IFNγ was required however, for the anti-tumor effectiveness of TC17 cells while IL-17 was not. This indicates that in the context of TC17 cell therapy, IFNγ is required for modulation of non-tumor cells in the tumor microenvironment, while IFNγ signaling in the tumor cells themselves is puzzlingly detrimental.
Within the first few days of adoptive transfer, TC1 cells proliferated faster in vivo than TC17 cells. However, after two and four weeks, in vivo levels of TC17 cells were higher or similar in the spleen and lungs compared with TC1 cells in mice bearing wild-type as well as IFNγ-nonresponsive tumors. Thus, the superior in vivo persistence of TC17 cells may be a factor in TC17 cell-elicited anti-tumor responses. IFNγ elicits its effects through activation of the STAT1 transcription factor, while IL-17 signals through an alternate ACT1 pathway to activate NF-ĸB. IL-22, also released by TC17 cells activates STAT3. Thus an interaction between these pathways in tumor cells may mediate the differential requirement for IFNγ-responsiveness in TC1 vs. TC17 mediated anti-tumor effects.
While it remains unclear why TC17 cells in this model were able to effectively control IFNγ-nonresponsive tumor cells but not wild-type tumor cells, and TC1 cells exhibited the opposite propensity, these observations have important implications for future enactment of adoptive cell transfer for tumor therapeutics. Because of their inherent stem-like propensity for long term in vivo persistence and demonstrations of highly effective anti-tumor functions, TH17 cells have been proposed to be a superior CD4+ cellular subset for adoptive anti-tumor T cell therapy in combinations with CD8+ T cells. However, the observations in this study suggest that different cytokine-producing CD8+ TC and CD4+ TH subsets will vary in their effectiveness depending on factors such as the tumor’s ability to respond to cytokines including IFNγ.
Further Reading:
Adoptive Transfer of Tc1 or Tc17 Cells Elicits Antitumor Immunity against Established Melanoma through Distinct Mechanisms. Yu Y, Cho HI, Wang D, Kaosaard K, Anasetti C, Celis E, Yu XZ. J Immunol. 2013 Feb 15;190(4):1873-81.
Tumor-specific Th17-polarized cells eradicate large established melanoma. Muranski P, Boni A, Antony PA, Cassard L, Irvine KR, Kaiser A, Paulos CM, Palmer DC, Touloukian CE, Ptak K, Gattinoni L, Wrzesinski C, Hinrichs CS, Kerstann KW, Feigenbaum L, Chan CC, Restifo NP. Blood. 2008 Jul 15;112(2):362-73.
Phenotype, distribution, generation, and functional and clinical relevance of Th17 cells in the human tumor environments. Kryczek I, Banerjee M, Cheng P, Vatan L, Szeliga W, Wei S, Huang E, Finlayson E, Simeone D, Welling TH, Chang A, Coukos G, Liu R, Zou W. Blood. 2009 Aug 6;114(6):1141-9.
Structure and signalling in the IL-17 receptor superfamily. Sarah L. Gaffen. Nat Rev Immunol. 2009 August; 9(8): 556.
Human TH17 cells are long-lived effector memory cells. Kryczek I, Zhao E, Liu Y, Wang Y, Vatan L, Szeliga W, Moyer J, Klimczak A, Lange A, Zou W. Sci Transl Med. 2011 Oct 12;3(104):104ra100.
Photo credit: AJC1 via photopin cc

 I previously posted about
I previously posted about 
 p53 (TP53), a transcription factor activated by various types of cellular stress, is one of the most well studied tumor suppressor proteins. Stress signals such as various types of DNA damage lead to p53 activation and subsequent induction of genes involved in cell cycle arrest, DNA repair, and apoptosis.
p53 (TP53), a transcription factor activated by various types of cellular stress, is one of the most well studied tumor suppressor proteins. Stress signals such as various types of DNA damage lead to p53 activation and subsequent induction of genes involved in cell cycle arrest, DNA repair, and apoptosis.
 Significant steps forward are being made in immunotherapeutic approaches for treatment of cancer. Over the past few years, two cancer immunotherapeutics were FDA approved. In 2010, Dendreon Corporation’s Provenge, an autologous cellular vaccine, was approved for hormone refractory metastatic prostate cancer. In 2011, Bristol-Myers Squibb’s anti-CTLA4 antibody Ipilimumab, was FDA approved for late stage melanoma. Recent promising clinical trial results indicate several additional immune modulating therapies are likely to join this prestigious list in the coming years. In addition, combinations of immune therapies with chemotherapeutics are being tested in clinical trials. In light of this, it is important to know how chemotherapies interact with the immune system, in order to best generate synergistic effects.
Significant steps forward are being made in immunotherapeutic approaches for treatment of cancer. Over the past few years, two cancer immunotherapeutics were FDA approved. In 2010, Dendreon Corporation’s Provenge, an autologous cellular vaccine, was approved for hormone refractory metastatic prostate cancer. In 2011, Bristol-Myers Squibb’s anti-CTLA4 antibody Ipilimumab, was FDA approved for late stage melanoma. Recent promising clinical trial results indicate several additional immune modulating therapies are likely to join this prestigious list in the coming years. In addition, combinations of immune therapies with chemotherapeutics are being tested in clinical trials. In light of this, it is important to know how chemotherapies interact with the immune system, in order to best generate synergistic effects.