Neurodegenerative diseases are characterized by usually fatal and progressive nervous system dysfunction caused by the death of neurons in the brain and spinal cord. In terms of human suffering and economic cost, neurodegenerative disorders carry an immense disease burden. However, despite extensive clinical research, especially in developing disease-modifying therapeutics, there is no effective medicine that halts or even slows any neurodegenerative disease. Currently in the United States, over 5 million Americans suffer from Alzheimer’s disease (AD), 1 million from Parkinson’s (PD), 400,000 from multiple sclerosis (MS), 30,000 from amyotrophic lateral sclerosis (ALS), and 30,000 from Huntington’s disease (HD). Thus, modification of current therapeutic research strategies and a more aggressive approach is a goal of increasing urgency.

Thus far, the majority of clinical research for treatment of neurodegenerative diseases has utilized disease-modifying therapeutics, which either prevent or target elimination of the pathogenetic causes or neurotoxins resulting from the disease. The basis for this approach revolves around several characteristics implicated in neurodegenerative diseases, such as accumulation of neurotoxic substances, autophagy and inflammation, as well as aggregation of misfolded proteins in neurodegenerative disorders, such as amyloid-β (Aβ) aggregates in AD or the mutant Huntington protein in HD, which take place prior to neuronal death. However, data obtained from several Phase III clinical trials indicate low efficacy of these treatments, specially in advanced stages of most neurodegenerative disorders; this is mainly due to the poor understanding of the underlying mechanisms of these disorders, hence the lack of knowledge of whether the targeted disease-characteristics are the cause or a symptom of the disease. Furthermore, the inability in early and accurate diagnosis of most neurodegenerative disorders impedes the early evaluation of therapeutic efficacy of new therapeutics.
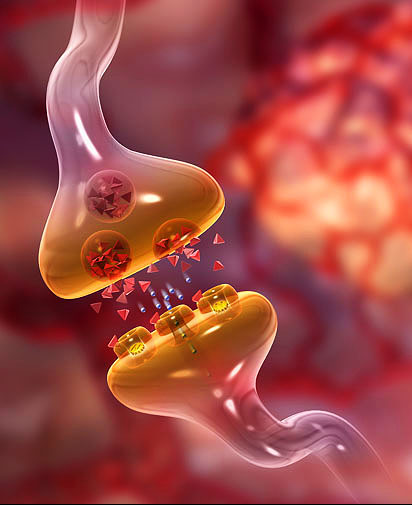
Although the underlying cellular processes contributing to HD, PD and AD differ, one common denominator in all these neurodegenerative diseases is the presence of inadequate neuronal communication, induced by the loss of synapses. Neuronal communication is carried out via synaptic transmission at neuronal synapses. A change in the properties of synaptic transmission due to brain’s ability to dynamically reorganize itself by forming new neuronal synapses is referred to as synaptic plasticity; compensation for injury as well as adjustment of neural activity in response to new stimuli or changes in their environment are among the most critical known functions of synaptic plasticity. Thus, degeneration of synapses leads to the loss of synaptic plasticity, preventing neuronal stimulation and eventual cell death.

According to a recent Review article published by scientists from GlaxoSmithKline in the advanced online edition of the Nature Reviews Neuroscience, Lu and colleagues present a compelling notion in treatment of neurodegenerative disorders; they propose a synaptic repair strategy targeting pathophysiology, which directly underlies the clinical syndromes. Unlike neuronal loss, synapse loss is reversible and synaptic dysfunction has the ability to be repaired, which allows the potential of neuronal repair prior to neuronal death. Furthermore, synaptic repair approach can be utilized for any neurological disease, regardless of the type or origin of the toxic byproduct. Lu’s group has proposed utilization of the synaptogenic molecule brain-derived neurotrophic factor (BDNF) as a potential synaptic repair therapeutic agent.
BDNF, an abundant neurotrophin expressed throughout the central nervous system, binds to NTRK2/TRKB and has been identified as one of the key neural signals regulating neuronal survival, neurogenesis and the only neurotrophin factor associated with synaptic plasticity in humans. Furthermore, in addition to its neuroprotective attribute, previous studies have demonstrated the key role of BDNF in cognitive functions, and synaptic deficit repair despite the presence of accumulated toxic proteins.
In this review, Lu’s team suggest potential routes of activating the BDNF pathway, as well as the importance of developing of a more reliable technique for quantification of synaptic changes in clinical settings, as essential tools in building effective disease-modifying therapies for neurodegenerative disorders. Although the notion of synaptic repair is an attractive one, the utilization of this strategy is still not feasible for the late stages of the disease, in which the irreversible neuronal death has already occurred. Furthermore, since most neurodegenerative diseases are diagnosed after the onset of neuronal death, emphasis on late stage treatment must remain a priority in neurodegenerative clinical research.
Further Reading:
BDNF-based synaptic repair as a disease-modifying strategy for neurodegenerative diseases





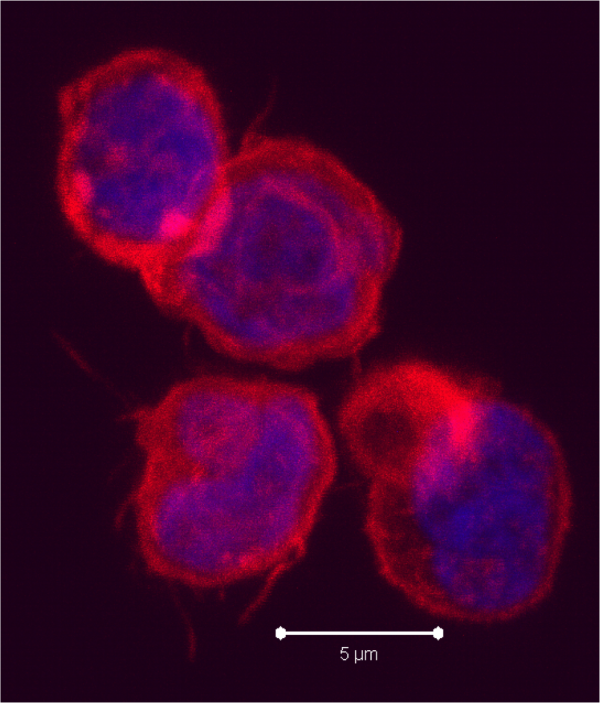





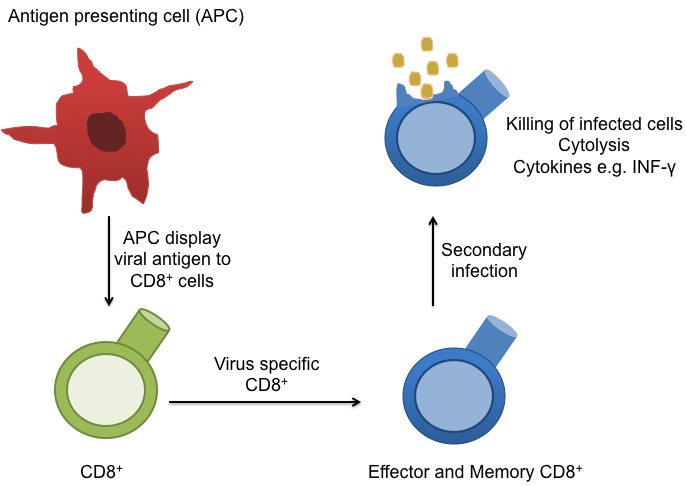
 circulate in the blood, lymph, and secondary lymphatic organs as they patrol for the presence of secondary infections. Recently, a class of effector memory CD8+ T cells has also been shown to migrate to and reside long-term in non-lymphoid tissues. These tissue resident memory T cells can quickly respond to tissue infections with their cognate pathogen. CD8+ T cells mainly function in killing of infected cells though cytolysis and by producing effector cytokines, including IFN-gamma to activate other immune cells. In a recent article in Nature Immunology,
circulate in the blood, lymph, and secondary lymphatic organs as they patrol for the presence of secondary infections. Recently, a class of effector memory CD8+ T cells has also been shown to migrate to and reside long-term in non-lymphoid tissues. These tissue resident memory T cells can quickly respond to tissue infections with their cognate pathogen. CD8+ T cells mainly function in killing of infected cells though cytolysis and by producing effector cytokines, including IFN-gamma to activate other immune cells. In a recent article in Nature Immunology,