
Human pluripotent stem cells (hPSCs) can differentiate into all cell types of the body, and can thereby serve as great models to examine the pathological mechanisms of various human diseases. At the International Society for Stem Cell Research (ISSCR) 11th Annual Meeting, various stem cell experts highlighted the current human stem cell models for Alzheimer’s disease, and discussed the potential future directions of the field.
Alzheimer’s disease (AD) is the most common neurodegenerative dementia, affecting ~30 million people worldwide. AD occurs in two main forms: early-onset, familial AD (FAD) and late-onset, sporadic AD (SAD). Both  are characterized by extensive neuronal loss and the aggregation of two proteins in the brain: amyloid β peptide (Aβ) and tau. Aβ peptide is derived from the amyloid precursor protein (APP) via cleavage by two proteases, β-secretase and γ-secretase. According to the amyloid cascade hypothesis, elevated levels of Aβ are necessary and sufficient to trigger disease 1. Tau is synthesized in neurons and normally functions in binding to tubulin and stabilization of microtubules. However, in AD, tau is hyper-phosphorylated, resulting in dissociation from microtubules, aggregation, and formation of neurofibrillary tangles (NFTs). Although the pathological hallmarks of AD consist of these amyloid plaques and NFTs, how the two are related to each other and how they contribute to clinical onset and progression of AD is still under investigation. By the time a patient manifests symptoms of a mild dementia, there is already significant neuronal loss and substantial accumulation of plaques and tangles. One major limitation to our understanding of AD has been the lack of live, patient-specific neurons to examine disease progression.
are characterized by extensive neuronal loss and the aggregation of two proteins in the brain: amyloid β peptide (Aβ) and tau. Aβ peptide is derived from the amyloid precursor protein (APP) via cleavage by two proteases, β-secretase and γ-secretase. According to the amyloid cascade hypothesis, elevated levels of Aβ are necessary and sufficient to trigger disease 1. Tau is synthesized in neurons and normally functions in binding to tubulin and stabilization of microtubules. However, in AD, tau is hyper-phosphorylated, resulting in dissociation from microtubules, aggregation, and formation of neurofibrillary tangles (NFTs). Although the pathological hallmarks of AD consist of these amyloid plaques and NFTs, how the two are related to each other and how they contribute to clinical onset and progression of AD is still under investigation. By the time a patient manifests symptoms of a mild dementia, there is already significant neuronal loss and substantial accumulation of plaques and tangles. One major limitation to our understanding of AD has been the lack of live, patient-specific neurons to examine disease progression.
With recent advances in reprogramming technology, scientists can now generate induced pluripotent stem cells (iPSCs), and thereby use live, patient-specific models to examine disease phenotypes in a dish. At the ISSCR meeting, Larry Goldstein presented his lab’s recent work on using hiPSC models to study AD. They generated iPSCs from two patients with FAD caused by a duplication of the APP gene, two patients with SAD, and two control individuals. Next, neurons were generated from the iPSC lines by directed differentiation and fluorescence-activated cell sorting (FACS) purification 2. Neurons from one SAD and two FAD patients demonstrated significantly higher levels of secreted Aβ and phosphorylated tau (p-tau) 3. To determine whether there is an association between APP processing and elevated p-tau levels, they treated iPSC-derived neurons with γ-secretase and β-secretase inhibitors. Interestingly, pharmacologic inhibition of β-secretase resulted in a significant reduction in the levels of Aβ and p-tau. Treatment with the γ-secretase inhibitor only reduced Aβ levels, but not p-tau levels. This suggests that products of APP processing other than Aβ might contribute to elevated p-tau levels, highlighting a potential weakness with the amyloid cascade hypothesis.
Other groups have proposed alternative hypotheses to explain AD pathogenesis. Haruhisa Inoue presented his group’s work on using human iPSC models to examine how intracellular Aβ oligomers contribute to AD. They generated iPSCs from one patient with FAD caused by the APP-E693Δ mutation, two patients with SAD, and three control individuals. Corticol neurons were derived using small molecule inhibitors of bone morphogenic protein (BMP) and activin/nodal signaling as previously described 4. Aβ oligomers accumulated in neurons derived from the FAD patient and one SAD patient, but not in the control neurons 5. Specifically, the Aβ oligomers accumulated in the endoplasmic reticulum (ER), and triggered ER and oxidative stress in the neurons. In addition, treatment with docosahexaenoic acid (DHA) alleviated the stress responses. Although the drug has previously failed in some clinical trials of AD treatment, Inoue’s work suggests that DHA might be effective for a subset of patients.
In summary, Goldstein and Inoue presented convincing evidence that human iPSC models can be used to study early AD pathogenesis and patient-specific drug responses. Although it can take decades for symptoms to manifest in patients, disease phenotypes can be observed using iPSC models. However, the fact that only one out of two SAD patients generated a disease phenotype highlights the need of future iPSC studies to examine larger numbers of patients to account for the observed heterogeneity in AD pathogenesis.
References:
1 Hardy, J. & Selkoe, D. J. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297, 353-356, doi:10.1126/science.1072994 (2002).
2 Yuan, S. H. et al. Cell-surface marker signatures for the isolation of neural stem cells, glia and neurons derived from human pluripotent stem cells. PLoS One 6, e17540, doi:10.1371/journal.pone.0017540 (2011).
3 Israel, M. A. et al. Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature 482, 216-220, doi:10.1038/nature10821 (2012).
4 Morizane, A., Doi, D., Kikuchi, T., Nishimura, K. & Takahashi, J. Small-molecule inhibitors of bone morphogenic protein and activin/nodal signals promote highly efficient neural induction from human pluripotent stem cells. J Neurosci Res 89, 117-126, doi:10.1002/jnr.22547 (2011).
5 Kondo, T. et al. Modeling Alzheimer’s disease with iPSCs reveals stress phenotypes associated with intracellular Abeta and differential drug responsiveness. Cell Stem Cell 12, 487-496, doi:10.1016/j.stem.2013.01.009 (2013).
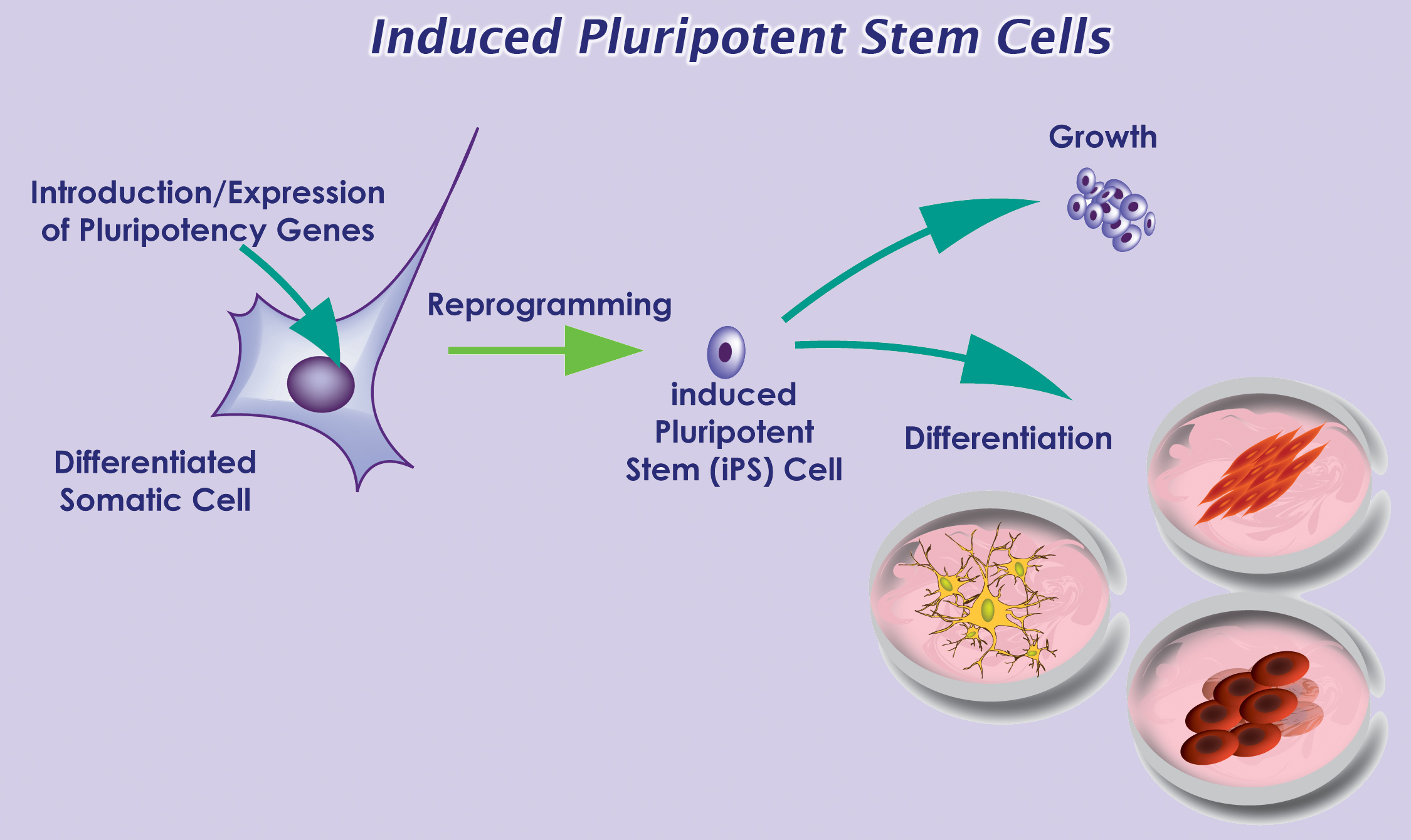






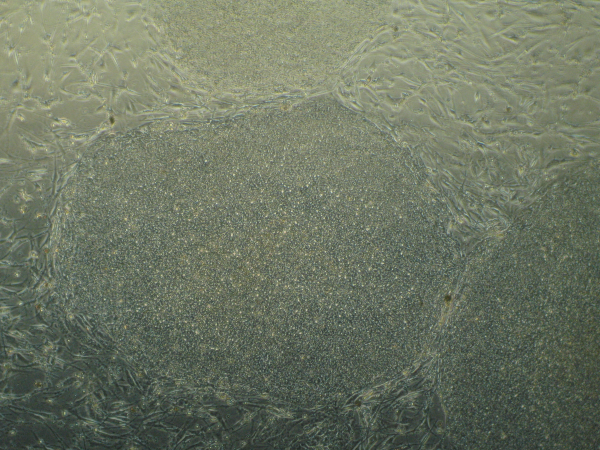
 Recently, in Stem Cells, Yamanaka’s group reported a protocol that increased the efficiency of iPSC induction from CD34+ cord blood and peripheral blood
Recently, in Stem Cells, Yamanaka’s group reported a protocol that increased the efficiency of iPSC induction from CD34+ cord blood and peripheral blood 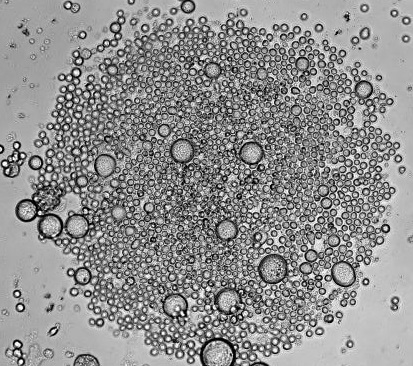
 Mature blood cells are relatively short-lived, and require replenishment from multipotent HSCs. Thus, HSCs must self-renew to generate an adequate pool of HSCs, as well as differentiate to give rise to more mature blood cells. A balance between self-renewal and differentiation ensures that the hematopoietic system can be functionally sustained throughout the lifetime of an adult body. However, as HSCs age, they accumulate DNA damage, often compromising their functionality. DNA damage can be further propagated both to daughter stem cells and downstream lineages, and may increase the risk of developing blood disorders
Mature blood cells are relatively short-lived, and require replenishment from multipotent HSCs. Thus, HSCs must self-renew to generate an adequate pool of HSCs, as well as differentiate to give rise to more mature blood cells. A balance between self-renewal and differentiation ensures that the hematopoietic system can be functionally sustained throughout the lifetime of an adult body. However, as HSCs age, they accumulate DNA damage, often compromising their functionality. DNA damage can be further propagated both to daughter stem cells and downstream lineages, and may increase the risk of developing blood disorders  However, exposure to ionizing radiation to induce myeloablation also causes DNA damage that can induce cell-cycle arrest or apoptosis of HSCs and their progenitor cells
However, exposure to ionizing radiation to induce myeloablation also causes DNA damage that can induce cell-cycle arrest or apoptosis of HSCs and their progenitor cells  In summary, EGF promotes HSC cycling and survival following radiation-induced myelosuppression. The study by Doan et al was the first demonstration that bone marrow HSCs express functional EGFR, and that EGFR signaling plays a role in HSC self-renewal. The results of this study suggest that EGF may have therapeutic potential to enhance hematopoietic regeneration in patients receiving myelosuppressive chemotherapy or undergoing HSC transplantation.
In summary, EGF promotes HSC cycling and survival following radiation-induced myelosuppression. The study by Doan et al was the first demonstration that bone marrow HSCs express functional EGFR, and that EGFR signaling plays a role in HSC self-renewal. The results of this study suggest that EGF may have therapeutic potential to enhance hematopoietic regeneration in patients receiving myelosuppressive chemotherapy or undergoing HSC transplantation.