
Melanoma, the most dangerous type of skin cancer, is the leading cause of death from skin disease (refer to my previous post titled “Targeting B-RAF in melanoma”). Melanoma results from a neoplastic transformation of pigment-producing melanocytes, with cutaneous neoplasms of stratified epithelium comprising the majority. Traditionally melanoma has been difficult to treat and exhibited resistance to all available standard therapies. However, recent progresses in the treatment of melanoma, including the FDA approved B-RAF inhibitor vemurafenib, have shown great promise in shrinking tumor size and improving the survival of patients. Studies have shown that melanoma is a complex disease that arises through multiple etiologic pathways. Therefore, detailed understanding of the underlying molecular mechanisms associated with melanoma pathogenesis and drug resistance is warranted in achieving a sustained clinical response. Over the past decade, molecular characterization of melanoma has progressed with the identification of the influence of various important oncogenes. These include oncogenic activation of BRAF, KIT, NRAS, cyclin D, and cyclin-dependent kinase 4 and alterations in the ERBB4 gene. Among these, the most frequently occurring genetic alteration associated with melanoma progression is the activating somatic mutation of B-RAF serine/thereonine kinase (for details please refer to my post titled “Targeting B-RAF in melanoma”). In addition to B-RAF mutations, which occurs in 50% of cases of melanoma, mutations of NRAS or neuroblastoma RAS gene were also identified in 15 to 20% of melanomas.

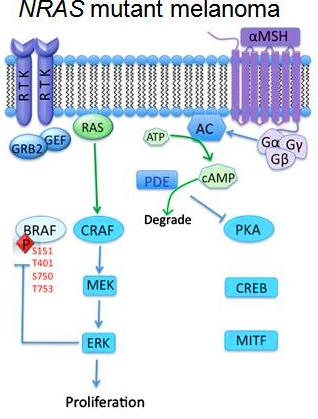
Approximately one-third of all human malignancies have mutations in RAS oncogene. RAS is a small sized plasma membrane-associated GTP binding protein. The RAS family of proteins consists of KRAS, HRAS, and NRAS. These proteins primarily regulate growth and, as a molecular switch, they connect signals from cell surface receptors to transcription factors and cell cycle regulating proteins in the nucleus. RAS proteins exist either in GTP-bound state (active) or GDP-bound (inactive) state. In normal cells, following binding of a ligand to its cognate receptor tyrosine kinase (RTK) RAS becomes activated. Once activated, RAS recruits and stimulates a number of signaling pathways including mitogen-activated protein kinas (MAPK) pathway and the phosphoinositide 3-kinase/AKT (PI3K/AKT) pathway.
Even though mutation of KRAS gene is the most common type of RAS mutation in human malignant disease, in melanoma, a point mutation of NRAS is most frequent and was the first oncogene to be identified. The most common mutation in NRAS is observed in codon 61 which occurs as a replacement of glutamine residue by lysine or arginine. This leads to constitutive activation of the MAPK signal transduction pathway resulting in proliferation and promotion of tumor growth. In addition, the RAS oncogene also activates signaling via the Rho GTPase Rac1, which can mediate growth, survival, and motility signaling.
Several studies analyzed the role of NRAS mutation in melanoma. In a study of 100 primary and metastatic melanoma samples by Ball et al. (1994), 36% of melanomas were found to contain mutations in RAS that, in 69% of cases, were at the codon 61. It was observed in multiple studies that patients with NRAS-mutated tumors are older at diagnosis than are patients with BRAF mutations (median age 55·7 years for NRAS vs 49·8 years for BRAF) and more frequently have melanoma due to chronic sun damage. Two large (>240 samples) studies of melanomas with NRAS mutations indicate that these tumors appear to exhibit more aggressive behavior, being associated with shorter overall survival. These tumors have also exhibited higher rates of mitosis and are thicker at presentation. A meta-analysis of studies from 1989 to 2010 reported that NRAS mutations were associated with nodular histology and location on the extremities. Collectively all these studies suggest a prominent role of NRAS as an oncogene in melanoma, and recommend scope of therapeutic targeting of NRAS for the treatment of advanced and high-risk melanoma.
With the increasing knowledge about the roles of various oncogenes (especially BRAF and NRAS), substantial advances in the targeted therapy to treat melanoma was achieved, however, only in BRAF-mutated melanomas. Compared to patients with BRAF mutations, as of today no approved targeted therapies exist for patients with NRAS-mutated melanoma. Complete inhibition of NRAS oncogenic signaling has proven to be challenging in part due to existence of redundant feedbacks to activate NRAS-MEK-ERK (MAPK) pathway. Various alternative strategies have thus been proposed, including (a) targeting membrane localization of RAS protein, required for RAS activity, through inhibitors of farnesyl transferase or galectin 1; (b) targeting NRAS mRNA with interfering RNAs; and (c) targeting signaling downstream of NRAS protein through inhibitors of PI3K/Akt. In addition, in in vitro studies some NRAS-mutated cell lines exhibited sensitivity to MEK inhibition. In conclusion, even though all these approaches hold promise, none of them got translated into the clinic. Therefore, more studies are required to formulate strategies to effectively treat NRAS-mutated melanoma.
References:
1.Devitt B, Liu W, Salemi R, Wolfe R, Kelly J, Tzen CY, Dobrovic A, McArthur G: Clinical outcome and pathological features associated with NRAS mutation in cutaneous melanoma. Pigment Cell Melanoma Res 2011, 24:666-672.
2. Ball NJ, Yohn JJ, Morelli JG, Norris DA, Golitz LE, Hoeffler JP: Ras mutations in human melanoma: a marker of malignant progression. J Invest Dermatol 1994, 102:285-290.
