We have all seen science fiction movies where alien invaders, in search of a fertile planet like earth, start a full scale war against humanity. A common theme in many of these stories, but often overlooked, is the role of the scout. Before any invasion, a solitary member of the invaders, fortified for the harsh journey, is sent on a quest to scout out the new land. As soon as this scout finds a promised land, the colony is formed and the invasion begins. This concept of invasion is seen in nature with viral and bacterial infections, where fortified virus or sporulated bacteria are able to survive harsh conditions and then proliferate in their host system upon arrival. In an ironic display, cancer metastasis follows a similar system; cancer cells are able to leave the primary tumor, travel long distances in harsh conditions, and form colonies in other tissues within an organism. These metastases have grave consequences. In fact in certain cancers, such as melanoma, it is metastasis to vital areas like the brain that makes it life-threatening. One of the biggest areas in cancer biology research is elucidating the mechanisms involved in cancer metastasis, which includes the concept of the epithelial to mesenchymal transition (EMT). It is this process that gives the cancer “scouts” the means to invade vasculature, fortify themselves for a journey to the metastatic site, and resist most therapies at all locations in the tumor-bearing patient. In normal development, the epithelial to mesenchymal transition is a reversible system that is involved in embryonic gastrulation1 and cardiac development1,2. In adulthood, EMT is involved in wound healing3. Unfortunately, EMT is also involved in pathological events such as fibrosis of injured tissue1 and cancer development and progression1-4. Here, we focus on EMT’s role in tumorigenesis, cancer proliferation, treatment resistance, and metastasis.
The epithelial to mesenchymal transition occurs in many different solid tumors of epithelial origin. Many tumors are carcinoma based, which implies that they are usually confined from motility by the basement membrane1 but the process of converting toward a cancer cell that has mesenchymal characteristics allows cancer cells to infiltrate the circulatory and lymphatic systems, causing motility that will later lead to metastasis1,5. As a cancer progresses, epithelial features, such as cellular adhesion marker E-cadherin3 and intracellular adhesion components such as tight junctions, cytokeratins, and desmosomes, are downregulated5. Coordinating with this downregulation is an upregulation of mesenchymal markers such as N-Cadherin, Vimentin, and Fibronectin3. This phenotypic shift is orchestrated by a coordination of many different mechanisms: epigenetic changes, post-translational modifications of proteins, transcriptional silencing by noncoding-RNAs (ncRNAs), and activation of epithelial to mesenchymal transcription factors (EMT-TFs)3. However, while all mechanisms of EMT development are important, this article will focus on the main orchestrators of EMT: the EMT-TFs.
EMT-TFs are many, but the main transcription factors fall into three families: SNAIL, ZEB, and TWIST. SNAIL has three main transcription factors: SNAIL (Snail1), SLUG (Snail2), and SMUC (Snail3)5. These proteins are zinc finger nucleases that are at the crux of EMT phenotype. In fact, it has been noted that SNAIL, and possibly SLUG, directly repress the expression of E-cadherin by binding to its promoter, CDH13. Furthermore, the SNAIL family has been shown to repress desmoplakin, adherens junctions, occulidins, and cytokeratin upon activation5. While not much is known about SMUC’s role in normal or pathological development, SNAIL, unlike SLUG, is so crucial to cancer metastasis that has been implicated in being an independent prognostic factor in the metastatic potential and severity of cancers5. Interestingly, not only does the SNAIL family propagate the characteristics of EMT transition, they also upregulate other EMT-TFs, such as ZEB1 and ZEB2, to further propagate the mechanism of EMT 5.
The Zinc-finger E-box-binding homeobox (ZEB) family are also a family of transcription factors with zinc-finger nuclease properties3. Like SNAIL, ZEB1 and ZEB2 bind to the E-cadherin promoter5. Moreover, ZEB1 and ZEB2 also downregulate tight/gap junctions, desmosomes and markers of polarity5. In addition, the ZEB family has been implicated in repressing P- and R-cadherins, other markers that inhibit the motility of epithelial cells5. ZEB1 is usually not found on non-cancerous cells but is found highly expressed on many cancer types5. In contrast, ZEB2 is expressed on normal epithelial cells, but is vastly upregulated in cancerous cells5.
 The final group is the TWIST family, a basic helix-loop-helix transcription factor involved in different steps in embryonic development1. Interestingly, while TWISTs are vital to embryogenesis, they are absent in normal adult epithelium5. As a cancerous cell progresses , TWIST1 and TWIST2 appear and their reactivity increases in correlation with the tumorigenic progress5. Interestingly, while TWIST1 is involved in regulating some of SLUG’s EMT effects, and directly drives expression of N-cadherin, it is not directly associated with the downregulation of E-cadherin5.
The final group is the TWIST family, a basic helix-loop-helix transcription factor involved in different steps in embryonic development1. Interestingly, while TWISTs are vital to embryogenesis, they are absent in normal adult epithelium5. As a cancerous cell progresses , TWIST1 and TWIST2 appear and their reactivity increases in correlation with the tumorigenic progress5. Interestingly, while TWIST1 is involved in regulating some of SLUG’s EMT effects, and directly drives expression of N-cadherin, it is not directly associated with the downregulation of E-cadherin5.
It is through these EMT-TFs that EMT is involved in tumorigenesis and tumor progression. EMT-TFs are involved in suppressing senescence and increasing cell cycle proliferation5. However, EMT-TFs are not enough to push tumorigenesis on their own, but require another event of tumorigenesis3. Therefore, EMT-TFs may act as a facilitator of tumorigenesis, but not be tumorigenic factors by themselves. EMT is also crucial for tumor invasiveness and metastasis. Besides the aforementioned changes in cellular adhesion molecules, activation of the EMT-TFs causes upregulation of matrix-metalloproteinases (MMP), enzymes involved in degradation of the extracellular matrix and invasion of cancer cells3. It has been demonstrated that the SNAIL family activates the expression of MMP1, MMP2, MMP7, and MT1-MP 5. Furthermore, the upregulation of MMPs additionally activates EMT-TFs, thus forming a feed-forward loop3. EMT events may also go beyond extracellular degradation; TWIST1 is known to be involved in the formation of invadopodia which has been correlated with invasiveness3.
The invasion of metastasis is not just due to intrinsic changes but also to changes in the host, and target microenvironment4. For instance, TGF-β, a cytokine that normally acts as a tumor suppressor, can enhance tumor invasion in later-stage tumors2. Furthermore, TGF-β activates the SNAIL and TWIST families of the EMT-TFs2,5. TGF-β may be produced by myeloid derived suppressor cells and CD11b+/F4/80+ tumor associated macrophages (TAMs) in the primary tumor microenvironment, thus perpetuating the EMT phenotype4. TAMs also express the cytokines fibroblast growth factor (FGF), epidermal growth factor (EGF), and macrophage colony stimulating factor (CSF-1) which are involved in EMT-based invasion as well as recruitment of immune cells toward a metastatic-favored microenvironment4. Moreover, TNF-α, usually correlated with an anti-tumor phenotype, also stabilizes SNAIL expression, thus implicating it in facilitating EMT3. Besides immune cells, other stromal cells are involved in EMT initiating and metastasis. For instance, mesenchymal stem cells (MSCs) and cancer associated fibroblasts (CAFs) are both involved in EMT initiation and propagation4.
One of the more clinically relevant aspects of EMT in cancer progression is the ability of EMT to confer therapy resistance. Resistance to doxorubicin is breast cancer correlates with higher levels of ZEB15 and both ZEB1 and ZEB2 have been shown to guard against cisplatin therapy5. TWIST1 mediates resistance to paciltaxel and TWIST1 and TWIST2 block daunorubicin by inhibiting degradation of the anti-apoptotic protein, Bcl-2, in bladder, ovarian, and prostate cancer5. Along the same lines, the EMT has been shown to confer upon cells a cancer stem cell-like phenotype2,4, a cancer phenotype also known for its therapy resistance5. TGF-β-driven EMT activates protein involved in stem cell phenotype, such as Sox2, PDGFB, and LIF 2. Furthermore, ZEB1 has been shown to be vital in the formation and maintenance of stem cell phenotype in some cancers5. However, while activation of the EMT pathway may confer a cancer stem cell-like phenotype, it is not necessary to get this phenotype3 as EMT-TFs are not necessarily involved in dedifferentiation3. In fact, some reports shine controversy on EMT-TFs’ role in stem cell development: while colorectal cancer spheroids show higher levels of SNAIL, other studies have shown that overexpression of SNAIL and SLUG in ovarian cancer drives these cancers away from a stem cell phenotype5.
 Besides the difficulty in treatment that EMT poses, one of the main obstacles that is troubling the cancer metastasis field is difficulty in identification of EMT/MET in cancer in vivo4. Because EMT is an orchestration between tumor and it’s microenvironment, researchers have been unable to definitively demonstrate the role of EMT beyond stromal epithelium4. The field needs better phenotypic markers to identify tumor epithelial cells from normal epithelial cells as well as a way to trace lineages of human cancers in vivo 4. Another is MET, the reverse of EMT and the end result of metastasis. To date, bona fide evidence for MET is only found in vitro studies and xenograft experiments4. MET explains why the phenotype of metastatic tumors mirror the primary site, but it is not an explanation for the system4. Several methods have been proposed to more appropriately study EMT, such as intravital 2-photon microscopy4. However, the sporadic nature of the EMT event makes observation, even in this system, very difficult4. In such a case, we are left to the spontaneous tumor-forming mouse models or studying xenograft models of immortalized cancer lines that are known to be highly metastatic. In addition, since there are no anatomically distinguishable between mesenchymal and epithelial cells, people have proposed the solutions of creating tumor lines the express reporter genes linked to promoters for epithelial/mesenchymal fates4. If one were able to combine an intravital two-photon system with a xenograft of a highly metastatic cancer line transduced with mesenchymal/epithelial reporter constructs, this would be the most feasible model to study EMT in real-time. As technology advances to detect individual cell populations in real-time, the expectation to solidify the mechanism of epithelial to mesenchymal transition in metastasis will increase the reliably of current EMT findings.
Besides the difficulty in treatment that EMT poses, one of the main obstacles that is troubling the cancer metastasis field is difficulty in identification of EMT/MET in cancer in vivo4. Because EMT is an orchestration between tumor and it’s microenvironment, researchers have been unable to definitively demonstrate the role of EMT beyond stromal epithelium4. The field needs better phenotypic markers to identify tumor epithelial cells from normal epithelial cells as well as a way to trace lineages of human cancers in vivo 4. Another is MET, the reverse of EMT and the end result of metastasis. To date, bona fide evidence for MET is only found in vitro studies and xenograft experiments4. MET explains why the phenotype of metastatic tumors mirror the primary site, but it is not an explanation for the system4. Several methods have been proposed to more appropriately study EMT, such as intravital 2-photon microscopy4. However, the sporadic nature of the EMT event makes observation, even in this system, very difficult4. In such a case, we are left to the spontaneous tumor-forming mouse models or studying xenograft models of immortalized cancer lines that are known to be highly metastatic. In addition, since there are no anatomically distinguishable between mesenchymal and epithelial cells, people have proposed the solutions of creating tumor lines the express reporter genes linked to promoters for epithelial/mesenchymal fates4. If one were able to combine an intravital two-photon system with a xenograft of a highly metastatic cancer line transduced with mesenchymal/epithelial reporter constructs, this would be the most feasible model to study EMT in real-time. As technology advances to detect individual cell populations in real-time, the expectation to solidify the mechanism of epithelial to mesenchymal transition in metastasis will increase the reliably of current EMT findings.
References:
1 Lim, J. & Thiery, J. P. Epithelial-mesenchymal transitions: insights from development. Development 139, 3471-3486, doi:10.1242/dev.071209 (2012).
2 Massagué, J. TGFβ signalling in context. Nat Rev Mol Cell Biol 13, 616-630, doi:10.1038/nrm3434 (2012).
3 Craene, B. D. & Berx, G. Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer 13, 97-110, doi:10.1038/nrc3447 (2012).
4 Gao, D., Vahdat, L. T., Wong, S., Chang, J. C. & Mittal, V. Microenvironmental regulation of epithelial-mesenchymal transitions in cancer. Cancer Res 72, 4883-4889, doi:10.1158/0008-5472.can-12-1223 (2012).
5 Sánchez-Tilló, E. et al. EMT-activating transcription factors in cancer: beyond EMT and tumor invasiveness. Cell Mol Life Sci 69, 3429-3456, doi:10.1007/s00018-012-1122-2 (2012).


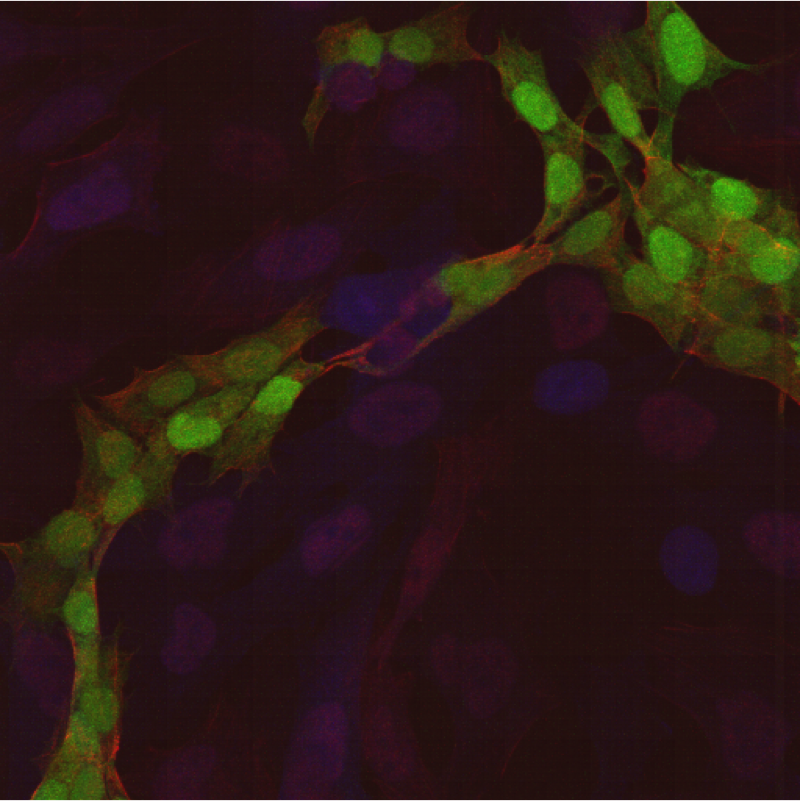


 CD95 (Fas, APO-1, TNFRSF6) is a member of the TNF-receptor superfamily and is best known for its role in mediating activation-induced cell death in activated T cells following binding to its ligand, CD95L/FasL induced on antigen-presenting cells (APCs). However, CD95 can also play additional, non-apoptotic roles in the modulation of T cell function. CD95 ligation has been shown to inhibit TCR signaling and activation of naïve T cells. However, this negative co-stimulatory effect appears to be dose-dependent, as low doses of CD95 agonists had the opposite effect and strongly promoted activation and proliferation of T cells. Like CD71, CD95 expression can be detected by 24 hours following T cell activation and continues to increase over the course of several days.
CD95 (Fas, APO-1, TNFRSF6) is a member of the TNF-receptor superfamily and is best known for its role in mediating activation-induced cell death in activated T cells following binding to its ligand, CD95L/FasL induced on antigen-presenting cells (APCs). However, CD95 can also play additional, non-apoptotic roles in the modulation of T cell function. CD95 ligation has been shown to inhibit TCR signaling and activation of naïve T cells. However, this negative co-stimulatory effect appears to be dose-dependent, as low doses of CD95 agonists had the opposite effect and strongly promoted activation and proliferation of T cells. Like CD71, CD95 expression can be detected by 24 hours following T cell activation and continues to increase over the course of several days.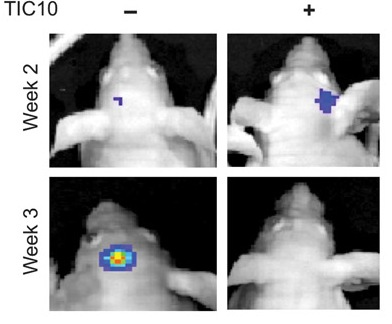
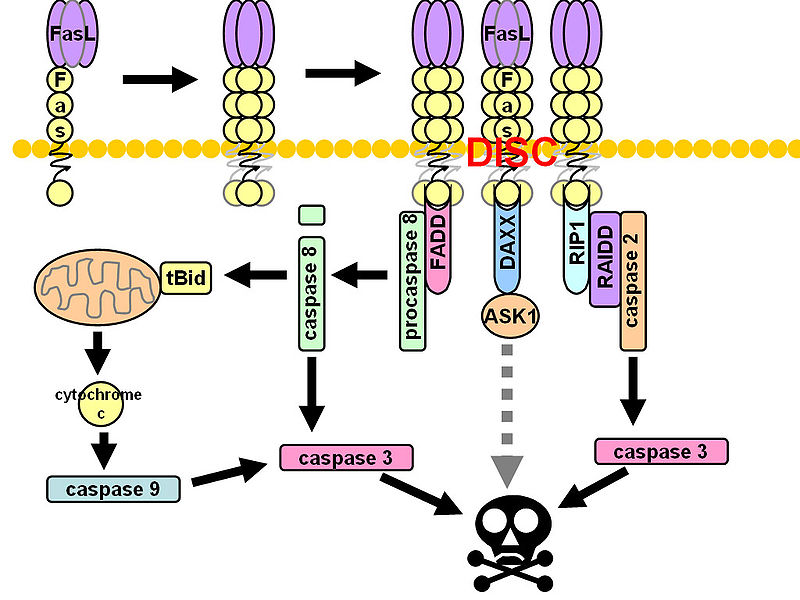
 R4) and TR2 (DR5). TRAIL also binds with two other receptors TR3 and TR4. Unlike TR1 and TR2, these receptors have incomplete death domains. Elevated expression of TR3 and TR4 in normal cells is thereby suggested to protect normal cells from TRAIL-induced death signaling. Induction of apoptotic signaling involves binding of TRAIL to DR4 or DR5. This results in homotrimerization and activation of receptors, enabling the receptors’ death domain to recruit the adaptor protein Fas-associated death domain along with pro-caspase- 8 and pro-caspase- 10. These all together then form the multi-protein death-inducing signaling complex, (DISC). Inside the DISC procaspeses become autoactivated and become caspase-8/10. Activated caspase-8/10 then activates downstream effector caspase-3 or caspase-7. Finally, cleavage of downstream substrates by effector caspases results in DNA fragmentation leading to apoptosis.
R4) and TR2 (DR5). TRAIL also binds with two other receptors TR3 and TR4. Unlike TR1 and TR2, these receptors have incomplete death domains. Elevated expression of TR3 and TR4 in normal cells is thereby suggested to protect normal cells from TRAIL-induced death signaling. Induction of apoptotic signaling involves binding of TRAIL to DR4 or DR5. This results in homotrimerization and activation of receptors, enabling the receptors’ death domain to recruit the adaptor protein Fas-associated death domain along with pro-caspase- 8 and pro-caspase- 10. These all together then form the multi-protein death-inducing signaling complex, (DISC). Inside the DISC procaspeses become autoactivated and become caspase-8/10. Activated caspase-8/10 then activates downstream effector caspase-3 or caspase-7. Finally, cleavage of downstream substrates by effector caspases results in DNA fragmentation leading to apoptosis. gen-activated protein kinase (MAPK) pathways was noted following TIC10 treatment in this in vivo study that resulted in translocation of the transcription factor Foxo3a into the nucleus, where Foxo3a induced expression of TRAIL gene to activate apoptosis.
gen-activated protein kinase (MAPK) pathways was noted following TIC10 treatment in this in vivo study that resulted in translocation of the transcription factor Foxo3a into the nucleus, where Foxo3a induced expression of TRAIL gene to activate apoptosis.



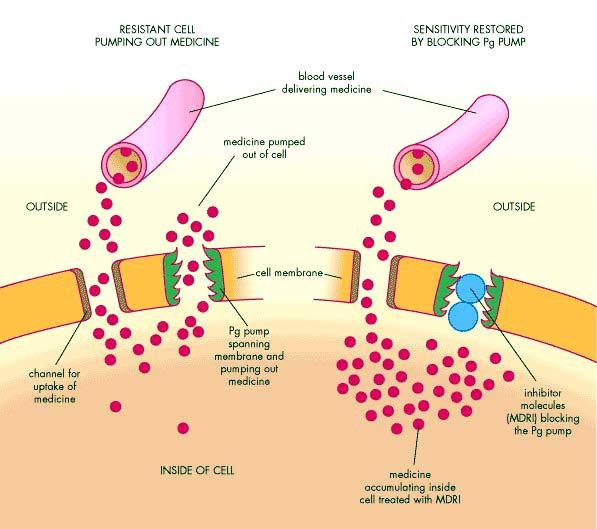
 As shown in Figure 1, tumor cells adopt several mechanisms to evade death induced by anti-tumor agents. These include changes in apoptotic pathways and activation of cell-cycle check points to increase DNA repair. Alternatively, cancer cells develop resistance by increased expression of multidrug-resistant proteins and altered anti-tumor drug transport mechanisms. Members of the ABC transporters (ATP-binding cassette) are known to be associated with this phenomenon, as the human genome express over 48 genes in this transporter family alone. These proteins bind ATP in their ATP binding domain and use the energy to transport various molecules across the cell, thus they are known as ABC proteins. Among these proteins, P-glycoprotein (Pgp, ABCB1), multidrug resistance-associated protein (MRP1, ABCC1), and breast cancer resistance protein (BCRP, ABCG2) are chiefly responsible for drug resistance in tumor cells. Studies are warranted to determine the role of other members of ABC transporters including MRP2, MRP3, MRP4, MRP5, ABCA2 and BSEP in drug-resistance.
As shown in Figure 1, tumor cells adopt several mechanisms to evade death induced by anti-tumor agents. These include changes in apoptotic pathways and activation of cell-cycle check points to increase DNA repair. Alternatively, cancer cells develop resistance by increased expression of multidrug-resistant proteins and altered anti-tumor drug transport mechanisms. Members of the ABC transporters (ATP-binding cassette) are known to be associated with this phenomenon, as the human genome express over 48 genes in this transporter family alone. These proteins bind ATP in their ATP binding domain and use the energy to transport various molecules across the cell, thus they are known as ABC proteins. Among these proteins, P-glycoprotein (Pgp, ABCB1), multidrug resistance-associated protein (MRP1, ABCC1), and breast cancer resistance protein (BCRP, ABCG2) are chiefly responsible for drug resistance in tumor cells. Studies are warranted to determine the role of other members of ABC transporters including MRP2, MRP3, MRP4, MRP5, ABCA2 and BSEP in drug-resistance. Plasma membrane glycoprotein (Pgp) was the first ABC-transporter detected in various cancers exerting resistance to a variety of chemically unrelated cytotoxic agents including anti-tumor drugs such as doxorubicin, vinblastine, ritonavir, indinavir and paclitaxel. It works as an energy-dependent efflux pump and can recognize a wide range of substrates. Even though this protein normally protects us from endogenous and exogenous toxins by transporting them out of the cells, the transporter causes a major problem in the bioavailability of anti-tumor drugs to tumor cells during chemotherapy.
Plasma membrane glycoprotein (Pgp) was the first ABC-transporter detected in various cancers exerting resistance to a variety of chemically unrelated cytotoxic agents including anti-tumor drugs such as doxorubicin, vinblastine, ritonavir, indinavir and paclitaxel. It works as an energy-dependent efflux pump and can recognize a wide range of substrates. Even though this protein normally protects us from endogenous and exogenous toxins by transporting them out of the cells, the transporter causes a major problem in the bioavailability of anti-tumor drugs to tumor cells during chemotherapy.
 I previously posted about
I previously posted about