

 In Part I, I talked about un-specific binding and Fc-receptor binding. Besides these cases of non-specific binding, there are also other cases of antibody/fluorochrome binding that appears non-specific but that actually represents a real specific interaction – even though it is usually one that is not welcomed. I call these ‘pseudo-artifacts’ and you will read about some really odd stuff here.
In Part I, I talked about un-specific binding and Fc-receptor binding. Besides these cases of non-specific binding, there are also other cases of antibody/fluorochrome binding that appears non-specific but that actually represents a real specific interaction – even though it is usually one that is not welcomed. I call these ‘pseudo-artifacts’ and you will read about some really odd stuff here.
(1) Binding of fluorochromes to Fc-receptors
The fact that Fc-receptors (FcR) bind antibodies is obvious, but lesser known is the fact that some of the fluorochrome linked to your antibody can also bind some FcRs.
It has been reported that R-phycoerythrin (PE) can bind to mouse Fc-gamma-RII (CD16) and Fc-gamma-RIII (CD32) (Takizawa et al.). Furthermore, FcR binding of fluorochromes apparently applies to most or maybe even all cyanine fluorochromes, either alone or in tandem conjugates (Shapiro). So far I found reports for Cy5 (Jahrsdorfer et al.), PE-Cy5 (van Vugt et al.; Steward and Steward; Jahrsdorfer et al.) and APC-Cy7 (Beavis et al.). In this case, human CD64 (Fc-gamma-RI) was suggested to be the culprit of some of the binding (van Vugt et al.; Jahrsdorfer et al.), but binding also to CD64neg leukemia cells has been reported (Steward and Steward), so the role of FcR is not solved for all cases yet.
Þ Potential solution:
(a) PE: For the binding of PE to mouse CD16/32 the use of a ‘Fc-block’, i.e. adding blocking monoclonal antibody 2.4G2 (rat IgG2b kappa), will avoid the problem (Takizawa et al.).
(b) Cyanine: If you work with FcR+ cells, especially monocytes, you might consider avoiding cyanine-containing fluorochromes for the staining of your cells of interest.
(2) Binding of fluorochromes to antigen-receptors
Phycoerythrin (PE) and allophycocyanin (APC) are large proteins of 240kD and 110kD respectively that were original derived from cyanobacteria or red algae. As it turns out these phycobiliproteins are also a specific antigen for some T and B cells. Approximately 0.1% of all mouse B cells recognize PE as antigen in a BCR-dependent manner (Pape et al.; Wu et al.). Similar, about 0.02% of all mouse B cells are APC antigen-specific (Pape et al.). Furthermore, about 0.02-0.4% of all gamma-delta-T cells (mouse and human) recognized PE as a specific antigen (Zeng et al.) as well.
Þ Potential solution: Given their low frequency, these cells only pose a problem if you study tiny subsets of B and gamma-delta-T cells. In that case, you should avoid the use of PE for your cells of interest.
(3) Binding of fluorochromes to other receptors or interaction partners
(a) Cross-reactivity of the antibody: Epitopes might be shared between different proteins, i.e. your antibody might not only recognize your protein in question, but recognizes also a similar epitope of another protein. This is for obvious reasons more likely with polyclonal antibodies.
Þ Potential solution: Usage of monoclonal antibodies reduces the risk of such cross-reactivity. If you suspect a cross-reactivity of your antibody, using a different clone for the same epitope will likely solve this problem.
(b) Intracellular biotin: Biotin is an important component of the cell metabolism. Therefore, biotin is present in the cells and the use of a streptavidin for intracellular staining will lead to binding of the streptavidin also to the cellular biotin.
Þ Potential solution: If you need to use a biotin-conjugated antibody for your intracellular staining you could cover all intracellular biotin by incubation of your cells with unconjugated streptavidin (followed by thorough washing) before the addition of your biotin-conjugated antibody.
(c) FITC charge: FITC is a charged molecule and antibodies with many FITC molecules (i.e. high F/P ratio) result in a highly charged antibody that binds, presumably through electrostatic interactions, nonspecifically to cytoplasmic elements (Hulspas et al.). This seems to be mainly a problem with intracellular staining and not with surface stains.
Þ Potential solution: For this reason FITC is not ideal for intracellular staining and you might try your antibody conjugated to a different fluorochrome.
(d) CD205: CD205 (DEC205) is a C-type lectin that is highly expressed on dendritic cells. Recently, it has been demonstrated that PE-Cy5.5 binds with high specificity to mouse CD205 (Park et al.). No staining was observed towards human CD205 and the binding of other Cy5.5 conjugates (PerCP-Cy5.5, APC-Cy5.5 and Cy5.5) to mouse CD205 was much weaker than that of PE-Cy5.5 (Park et al.).
Þ Potential solution: Given the high specificity of the interaction, you should avoid the use of PE-Cy5.5, and to a lesser extent other Cy5.5 containing fluorochromes, when your cells of interest expresses mouse CD205.
(4) Other effects
Finally, another odd-ball has been reported for APC tandems. Apparently, living cells have some way, which depends on their metabolism, to degrade the APC-Cy7 and APC-H7 tandems, leaving you with an APC signal (Le Roy et al.). APC-Cy7 seemed to be more affected than APC-H7 and monocytes are more active at degrading this signal than lymphocytes.
Þ Potential solution: Given that this requires live cells, fixation of your cell solution after staining will solve this problem. Alternatively, as this degradation requires metabolically active cells, storing your cell solution at 4°C or on ice or adding sodium azide (NaN3) to your storing buffer will reduce the effect.
That’s all I have for now, but if you know of other such ‘pseudo-artifacts’, or if you have any corrections and comments please share them with us!

//
References:
An amazing source for odd questions on flow cytometry is the ‘Cytometry mailing list’ hosted by the Purdue University, which can be found under: https://lists.purdue.edu/mailman/listinfo/cytometry
Beavis, A.J. & Pennline, K.J., 1996. Allo-7: a new fluorescent tandem dye for use in flow cytometry. Cytometry, 24(4), pp.390–395.
Hulspas, R. et al., 2009. Considerations for the control of background fluorescence in clinical flow cytometry. Cytometry, 76B(6), pp.355–364.
Jahrsdörfer, B., Blackwell, S.E. & Weiner, G.J., 2005. Phosphorothyoate oligodeoxynucleotides block nonspecific binding of Cy5 conjugates to monocytes,
Le Roy, C. et al., 2009. Flow cytometry APC-tandem dyes are degraded through a cell-dependent mechanism. Cytometry A, 75(10), pp.882–890.
Pape, K.A. et al., 2011. Different B cell populations mediate early and late memory during an endogenous immune response. Science, 331(6021), pp.1203–1207.
Park, C.G., Rodriguez, A. & Steinman, R.M., 2012. PE-Cy5.5 conjugates bind to the cells expressing mouse DEC205/CD205. J Immunol Methods, 384(1-2), pp.184–190.
Shapiro, H.M., 2004. Practical Flow Cytometry 4th Edt,
Stewart, C.C. & Stewart, S.J., 2001. Cell preparation for the identification of leukocytes. Methods Cell Biol, 63, pp.217–251.
Takizawa, F., Kinet, J.P. & Adamczewski, M., 1993. Binding of phycoerythrin and its conjugates to murine low affinity receptors for immunoglobulin G. Journal of Immunological Methods, 162(2), pp.269–272.
van Vugt, M.J., van den Herik-Oudijk, I.E. & van de Winkle, J.G., 1996. Binding of PE-CY5 conjugates to the human high-affinity receptor for IgG (CD64). Blood, 88(6), pp.2358–2361.
Wu, C.J. et al., 1991. Murine memory B cells are multi-isotype expressors. Immunology, 72(1), pp.48–55.
Zeng, X. et al., 2012. gd T Cells Recognize a Microbial Encoded B Cell Antigen to Initiate a Rapid Antigen-Specific Interleukin-17 Response. Immunity, 37(3), pp.524–534.
 Gerhard Wingender is currently an Instructor at the La Jolla Institute for Allergy and Immunology (La Jolla, CA). His main lab toy is flow cytometry and his research interest involve invariant Natural Killer T (iNKT) cells.
Gerhard Wingender is currently an Instructor at the La Jolla Institute for Allergy and Immunology (La Jolla, CA). His main lab toy is flow cytometry and his research interest involve invariant Natural Killer T (iNKT) cells.

//
Photo credit: PNNL – Pacific Northwest National Laboratory / Foter.com / CC BY-NC-SA

 CXCR5 is a chemokine receptor expressed by and used to identity human CD4+ follicular helper T cells (TFH). TFH cells, as their name implies, promote the differentiation and survival of memory and plasma B cells in the B cell follicular and germinal center regions of secondary lymphoid organs. CXCR5+ TFH-like central memory CD4+ T cells (CD4+ TCM) also circulate in peripheral blood and can be detected among human peripheral blood mononuclear cells (PBMC). CXCR5+ cells comprise 20-25% of CD4+ TCM cells in human PBMC. However, what are the identifying markers and functional differences of CXCR5+ vs. CXCR5– CD4+ T cells from human PBMC and the prototypical CXCR5+ TFH cells in secondary lymphoid organs?
CXCR5 is a chemokine receptor expressed by and used to identity human CD4+ follicular helper T cells (TFH). TFH cells, as their name implies, promote the differentiation and survival of memory and plasma B cells in the B cell follicular and germinal center regions of secondary lymphoid organs. CXCR5+ TFH-like central memory CD4+ T cells (CD4+ TCM) also circulate in peripheral blood and can be detected among human peripheral blood mononuclear cells (PBMC). CXCR5+ cells comprise 20-25% of CD4+ TCM cells in human PBMC. However, what are the identifying markers and functional differences of CXCR5+ vs. CXCR5– CD4+ T cells from human PBMC and the prototypical CXCR5+ TFH cells in secondary lymphoid organs?
 In this first half of the two part blog, I will talk about the two reasons, namely unspecific binding and Fc-receptors, which most people think of when they talk about non-specific binding in flow cytometry.
In this first half of the two part blog, I will talk about the two reasons, namely unspecific binding and Fc-receptors, which most people think of when they talk about non-specific binding in flow cytometry.


 Arijit
Arijit
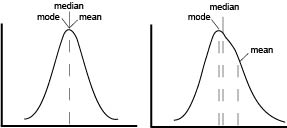
 The first point of confusion is born from the name itself. MFI is often used without explanation, to abbreviate either arithmetic mean, geometric mean, or median fluorescence intensity. In a perfect world, our data would be normally distributed and in that case means, median and mode are all equal. In reality, flow data is rarely normal and never perfect. The more that the data skews, the further the mean drifts in the direction of skew and becomes less representative of the data being analyze as seen on the graphical representation.
The first point of confusion is born from the name itself. MFI is often used without explanation, to abbreviate either arithmetic mean, geometric mean, or median fluorescence intensity. In a perfect world, our data would be normally distributed and in that case means, median and mode are all equal. In reality, flow data is rarely normal and never perfect. The more that the data skews, the further the mean drifts in the direction of skew and becomes less representative of the data being analyze as seen on the graphical representation. Adam
Adam
 Natural Killer (NK) cells are a cytotoxic innate immune lymphocyte cell type. In humans, NK cells comprise up to 15% of peripheral blood mononuclear cells (PBMC), and 5-20% of the PBMC lymphocyte population. Several subtypes of NK cells exist in humans. In this post, I will discuss phenotypic properties and markers of NK subtypes present in human PBMC.
Natural Killer (NK) cells are a cytotoxic innate immune lymphocyte cell type. In humans, NK cells comprise up to 15% of peripheral blood mononuclear cells (PBMC), and 5-20% of the PBMC lymphocyte population. Several subtypes of NK cells exist in humans. In this post, I will discuss phenotypic properties and markers of NK subtypes present in human PBMC.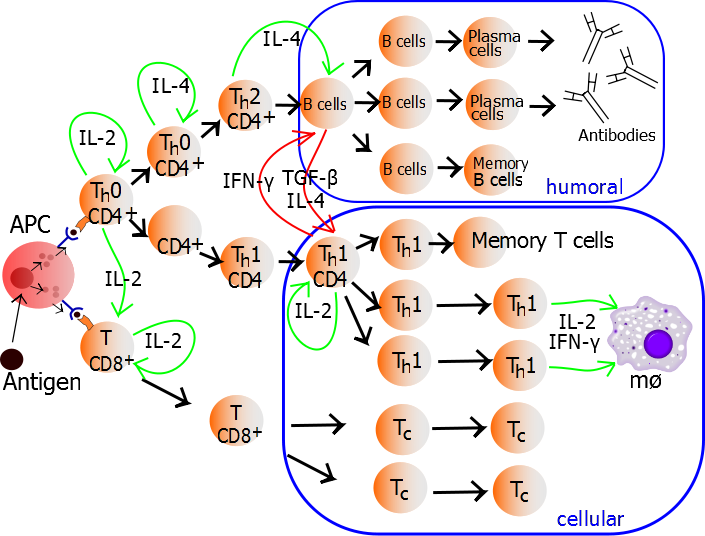
 TH1 cells can be generated and assayed for functions including IFNγ expression in as few as three days. If long term or clonal T cells assays are of interest, cells can be expanded in the presence of IL-2 for 2-3 weeks following single cell cloning.
TH1 cells can be generated and assayed for functions including IFNγ expression in as few as three days. If long term or clonal T cells assays are of interest, cells can be expanded in the presence of IL-2 for 2-3 weeks following single cell cloning. 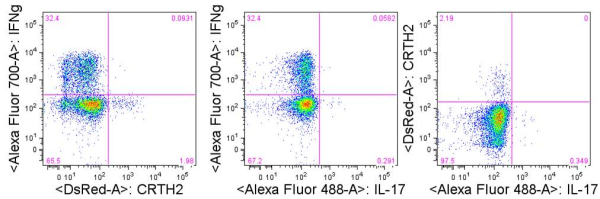

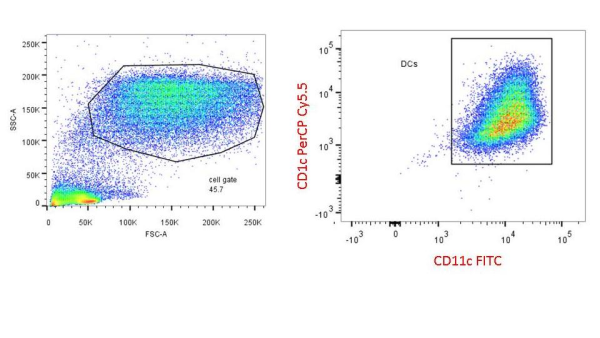

 Colt Egelston is currently a post-doctoral fellow at the Beckman Research Institute of the City of Hope, in Duarte, CA. He received his Ph.D. from Rush University in Chicago and is interested in all things immunology.
Colt Egelston is currently a post-doctoral fellow at the Beckman Research Institute of the City of Hope, in Duarte, CA. He received his Ph.D. from Rush University in Chicago and is interested in all things immunology.
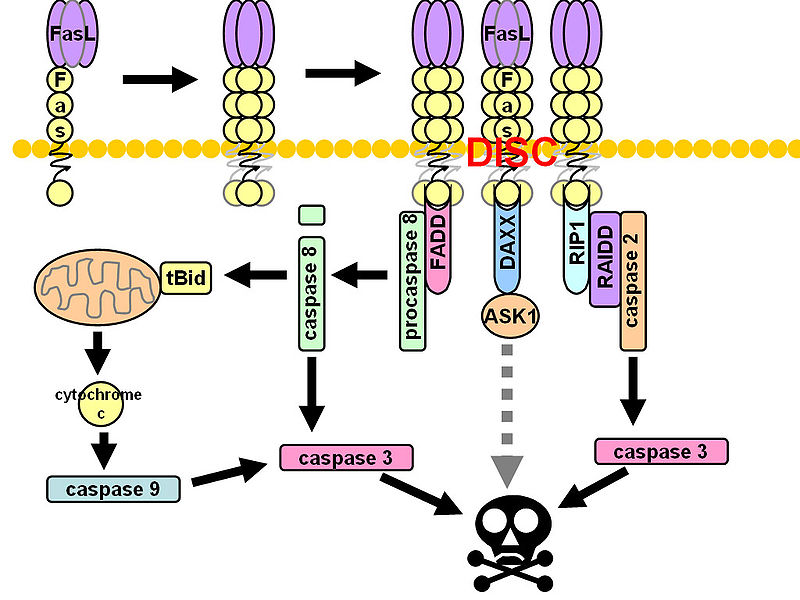
 CD95 (Fas, APO-1, TNFRSF6) is a member of the TNF-receptor superfamily and is best known for its role in mediating activation-induced cell death in activated T cells following binding to its ligand, CD95L/FasL induced on antigen-presenting cells (APCs). However, CD95 can also play additional, non-apoptotic roles in the modulation of T cell function. CD95 ligation has been shown to inhibit TCR signaling and activation of naïve T cells. However, this negative co-stimulatory effect appears to be dose-dependent, as low doses of CD95 agonists had the opposite effect and strongly promoted activation and proliferation of T cells. Like CD71, CD95 expression can be detected by 24 hours following T cell activation and continues to increase over the course of several days.
CD95 (Fas, APO-1, TNFRSF6) is a member of the TNF-receptor superfamily and is best known for its role in mediating activation-induced cell death in activated T cells following binding to its ligand, CD95L/FasL induced on antigen-presenting cells (APCs). However, CD95 can also play additional, non-apoptotic roles in the modulation of T cell function. CD95 ligation has been shown to inhibit TCR signaling and activation of naïve T cells. However, this negative co-stimulatory effect appears to be dose-dependent, as low doses of CD95 agonists had the opposite effect and strongly promoted activation and proliferation of T cells. Like CD71, CD95 expression can be detected by 24 hours following T cell activation and continues to increase over the course of several days.